Comparative Analysis of Single Cell Transcriptomes of Human Blastoids
Jialei Duan 
Human blastoids provide a readily accessible, scalable, versatile and perturbable alternative to blastocysts for studying early human development, understanding early pregnancy loss and gaining insights into early developmental defects.
In this manuscript, several transcriptome data sets generated by different technologies were included. To minimize platform and processing differences, raw fastq files of public datasets using the Illumina sequencing platform were downloaded and re-processed.
- Cinkornpumin, J.K., Kwon, S.Y., Guo, Y., Hossain, I., Sirois, J., Russett, C.S., Tseng, H.-W., Okae, H., Arima, T., Duchaine, T.F., et al. (2020). Naive human embryonic stem cells can give rise to cells with a trophoblast-like transcriptome and methylome. Stem Cell Reports 15, 198–213.
- Dong, C., Beltcheva, M., Gontarz, P., Zhang, B., Popli, P., Fischer, L.A., Khan, S.A., Park, K.-M., Yoon, E.-J., Xing, X., et al. (2020). Derivation of trophoblast stem cells from naïve human pluripotent stem cells. Elife 9.
- Linneberg-Agerholm, M., Wong, Y.F., Romero Herrera, J.A., Monteiro, R.S., Anderson, K.G.V., and Brickman, J.M. (2019). Naïve human pluripotent stem cells respond to Wnt, Nodal and LIF signalling to produce expandable naïve extra-embryonic endoderm. Development 146.
- Lv, B., An, Q., Zeng, Q., Zhang, X., Lu, P., Wang, Y., Zhu, X., Ji, Y., Fan, G., and Xue, Z. (2019). Single-cell RNA sequencing reveals regulatory mechanism for trophoblast cell-fate divergence in human peri-implantation conceptuses. PLoS Biol. 17, e3000187.
- Nakamura, T., Okamoto, I., Sasaki, K., Yabuta, Y., Iwatani, C., Tsuchiya, H., Seita, Y., Nakamura, S., Yamamoto, T., and Saitou, M. (2016). A developmental coordinate of pluripotency among mice, monkeys and humans. Nature 537, 57–62.
- Petropoulos, S., Edsgärd, D., Reinius, B., Deng, Q., Panula, S.P., Codeluppi, S., Plaza Reyes, A., Linnarsson, S., Sandberg, R., and Lanner, F. (2016). Single-cell RNA-Seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell 165, 1012–1026.
- Takashima, Y., Guo, G., Loos, R., Nichols, J., Ficz, G., Krueger, F., Oxley, D., Santos, F., Clarke, J., Mansfield, W., et al. (2014). Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 158, 1254–1269.
- Xiang, L., Yin, Y., Zheng, Y., Ma, Y., Li, Y., Zhao, Z., Guo, J., Ai, Z., Niu, Y., Duan, K., et al. (2020). A developmental landscape of 3D-cultured human pre-gastrulation embryos. Nature 577, 537–542.
- Zhou, F., Wang, R., Yuan, P., Ren, Y., Mao, Y., Li, R., Lian, Y., Li, J., Wen, L., Yan, L., et al. (2019). Reconstituting the transcriptome and DNA methylome landscapes of human implantation. Nature 572, 660–664.
Sys.time()[1] "2022-09-25 00:35:30 CDT"[1] "America/Chicago"Preparation
Functions
Load required packages.
source(
file = file.path(
SCRIPT_DIR,
"utilities.R"
)
)
load_matrix <- function(x) {
matrix_readcount_use <- scipy$sparse$load_npz(
file.path(x, "matrix_readcount.npz")
)
colnames(matrix_readcount_use) <- np$load(
file.path(x, "matrix_readcount_barcodes.npy")
)
rownames(matrix_readcount_use) <- np$load(
file.path(x, "matrix_readcount_features.npy")
)
return(matrix_readcount_use)
}
perform_pca <- function(matrix, features, min_counts = 1) {
matrix_norm <- matrix[
Matrix::rowSums(
matrix
) >= min_counts,
]
matrix_norm <- as(matrix_norm, "dgCMatrix")
matrix_norm@x <- median(colSums(matrix_norm)) *
(matrix_norm@x / rep.int(
colSums(matrix_norm),
diff(matrix_norm@p)
))
matrix_norm_log <- matrix_norm[
rownames(matrix_norm) %in% features,
]
matrix_norm_log@x <- log1p(matrix_norm_log@x)
# z-score
matrix_norm_log_scaled <- t(
scale(
t(
matrix_norm_log
),
center = TRUE, scale = TRUE
)
)
# PCA
pca_out <- prcomp(
t(matrix_norm_log_scaled),
center = FALSE,
scale = FALSE
)
return(pca_out)
}Python env
reticulate::py_config()python: /Users/jialei/.pyenv/shims/python
libpython: /Users/jialei/.pyenv/versions/mambaforge-4.10.3-10/lib/libpython3.9.dylib
pythonhome: /Users/jialei/.pyenv/versions/mambaforge-4.10.3-10:/Users/jialei/.pyenv/versions/mambaforge-4.10.3-10
version: 3.9.13 | packaged by conda-forge | (main, May 27 2022, 17:00:33) [Clang 13.0.1 ]
numpy: /Users/jialei/.pyenv/versions/mambaforge-4.10.3-10/lib/python3.9/site-packages/numpy
numpy_version: 1.22.4
numpy: /Users/jialei/.pyenv/versions/mambaforge-4.10.3-10/lib/python3.9/site-packages/numpy
NOTE: Python version was forced by RETICULATE_PYTHONPROJECT_DIR <- "/Users/jialei/Dropbox/Data/Projects/UTSW/Human_blastoid"Matrix
MATRIX_DIR <- list(
"github/data/matrices/LW36",
"github/data/matrices/LW49_LW50_LW51_LW52",
"github/data/matrices/LW58_LW59",
"github/data/matrices/LW60_LW61",
"raw/public/PRJEB11202/reformatted_matrix"
)
matrix_readcount_use <- purrr::map(MATRIX_DIR, \(x) {
load_matrix(file.path(PROJECT_DIR, x))
}) |>
purrr::reduce(cbind)
# clean up
rm(MATRIX_DIR)Embedding
Metadata
cell_metadata_PRJEB11202 <- read_delim(
file.path(
PROJECT_DIR,
"raw/public/PRJEB11202/",
"E-MTAB-3929.sdrf.tsv"
),
delim = "\t"
) |>
dplyr::select(
`Comment[ENA_SAMPLE]`,
`Comment[ENA_RUN]`,
`Characteristics[developmental stage]`,
`Characteristics[inferred lineage]`
) |>
dplyr::rename(
cell = `Comment[ENA_SAMPLE]`,
run = `Comment[ENA_RUN]`,
developmental_stage = `Characteristics[developmental stage]`,
lineage = `Characteristics[inferred lineage]`
) |>
dplyr::mutate(
developmental_stage = stringr::str_replace(
string = developmental_stage,
pattern = "embryonic day ",
replacement = "E"
),
lineage = dplyr::case_when(
lineage == "epiblast" ~ "EPI",
lineage == "primitive endoderm" ~ "HYP",
lineage == "trophectoderm" ~ "TE",
lineage == "not applicable" ~ "Pre-lineage"
)
)
embedding <- embedding |>
dplyr::left_join(
cell_metadata_PRJEB11202
) |>
dplyr::mutate(
lineage = dplyr::case_when(
is.na(lineage) ~ "Blastoid",
TRUE ~ as.character(lineage)
),
developmental_stage = dplyr::case_when(
is.na(developmental_stage) ~ "Blastoid",
TRUE ~ as.character(developmental_stage)
)
)
walk(list(matrix_readcount_use), \(x) {
print(object.size(x), units = "auto", standard = "SI")
})1.6 GBPre/post-implantation comparison
Blastoid
matrix_readcount_blastoid_pseudobulk <- purrr::map(
sort(unique(embedding$louvain)), \(x) {
cells_in_group <- embedding |>
dplyr::filter(
louvain == x,
batch != "PRJEB11202"
) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_use[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(sort(unique(embedding$louvain)))EMBEDDING_FILE <- "embedding_stem.csv.gz"
embedding_stem <- read_csv(
file = file.path(
PROJECT_DIR,
"github",
"data",
EMBEDDING_FILE
)
)
matrix_readcount_stem_pseudobulk <- purrr::map(
sort(unique(embedding_stem$batch)), \(x) {
cells_in_group <- embedding_stem |>
dplyr::filter(batch == x) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_use[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(sort(unique(embedding_stem$batch)))Public datasets
Petropoulos et al
Single-cell RNA-Seq reveals lineage and X chromosome dynamics in human preimplantation embryos
- BioProject Accession: PRJEB11202
- ArrayExpress: E-MTAB-3929
- Notebook: https://jlduan.github.io/replica/j.cell.2016.03.023
matrix_readcount_PRJEB11202_pseudobulk_EPI <- purrr::map(
c("E5", "E6", "E7"), \(x) {
cells_in_group <- embedding |>
dplyr::filter(
lineage == "EPI",
developmental_stage == x
) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_use[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(c("E5", "E6", "E7")) |>
as.matrix()
matrix_readcount_PRJEB11202_pseudobulk_TE <- purrr::map(
c("E5", "E6", "E7"), \(x) {
cells_in_group <- embedding |>
dplyr::filter(
lineage == "TE",
developmental_stage == x
) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_use[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(c("E5", "E6", "E7")) |>
as.matrix()
matrix_readcount_PRJEB11202_pseudobulk_HYP <- purrr::map(
c("E5", "E6", "E7"), \(x) {
cells_in_group <- embedding |>
dplyr::filter(
lineage == "HYP",
developmental_stage == x
) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_use[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(c("E5", "E6", "E7")) |>
as.matrix()Xiang et al
A developmental landscape of 3D-cultured human pre-gastrulation embryos
- BioProject Accession: PRJNA562548
- GEO Accession: GSE136447
- Notebook: https://jlduan.github.io/replica/s41586-019-1875-y
cell_metadata_PRJNA562548 <- vroom::vroom(
file = file.path(
MATRIX_DIR, "cell_metadata.csv"
)
) |>
dplyr::select(
cell = `Sample Name`,
developmental_stage = Age,
lineage = Cell_type,
run = Run
) |>
dplyr::mutate(
developmental_stage = stringr::str_replace(
string = developmental_stage,
pattern = "embryo invitro day ",
replacement = "E"
),
developmental_stage = factor(
developmental_stage,
levels = stringr::str_sort(
x = unique(developmental_stage),
numeric = TRUE
)
),
lineage = factor(
lineage,
levels = c(
"ICM",
"EPI",
"PSA-EPI",
"Hypoblast",
"CTBs",
"STBs",
"EVTs"
)
)
)matrix_readcount_PRJNA562548_pseudobulk_EPI <- purrr::map(
levels(cell_metadata_PRJNA562548$developmental_stage), \(x) {
cells_in_group <- cell_metadata_PRJNA562548 |>
dplyr::filter(
lineage == "EPI",
developmental_stage == x
) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_PRJNA562548[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(levels(cell_metadata_PRJNA562548$developmental_stage)) |>
as.matrix()
matrix_readcount_PRJNA562548_pseudobulk_HYP <- purrr::map(
levels(cell_metadata_PRJNA562548$developmental_stage), \(x) {
cells_in_group <- cell_metadata_PRJNA562548 |>
dplyr::filter(
lineage == "Hypoblast",
developmental_stage == x
) |>
dplyr::pull(cell)
if (length(cells_in_group) > 1) {
m <- Matrix::rowSums(matrix_readcount_PRJNA562548[, cells_in_group])
} else {
m <- matrix_readcount_PRJNA562548[, cells_in_group]
}
return(m)
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(
cell_metadata_PRJNA562548 |>
dplyr::filter(lineage == "Hypoblast") |>
dplyr::pull(developmental_stage) |>
droplevels() |>
levels()
) |>
as.matrix()
# clean up
rm(matrix_readcount_PRJNA562548)
rm(cell_metadata_PRJNA562548)Zhou et al
Reconstituting the transcriptome and DNA methylome landscapes of human implantation
- BioProject Accession: PRJNA431392
- GEO Accession: GSE109555
- Notebook: https://jlduan.github.io/replica/s41586-019-1500-0
embryos_selected_PRJNA431392 <- c(
"ha_D6_E2",
"hm_D6_E1",
"hm_D6_E2",
"hm_D8_E2",
"hm_D8_E3",
"hm_D8_E5",
"ha_D8_E1",
"hm_D8_E1",
"hv_D8_E1",
"hv_D8_E2",
"hv_D8_E3",
"hv_D10_E6",
"ha_D10_E1",
"ha_D10_E2",
"hm_D10_E4",
"hm_D10_E9",
"hv_D10_E7",
"hv_D10_E8",
"ha_D12_E1",
"hv_D12_E1",
"hv_D12_E2"
)
cell_metadata_PRJNA431392 <- vroom::vroom(
file = file.path(
MATRIX_DIR, "cell_metadata.csv"
)
) |>
dplyr::mutate(
developmental_stage = str_replace(
string = Day,
pattern = "D",
replacement = "E"
),
developmental_stage = factor(
developmental_stage,
levels = stringr::str_sort(
unique(developmental_stage),
numeric = TRUE
)
),
lineage = factor(
Lineage,
levels = c(
"EPI",
"PE",
"TE",
"MIX"
)
),
`3184` = case_when(
Ori_Day_Emb %in% embryos_selected_PRJNA431392 ~ "1",
TRUE ~ "0"
)
) |>
dplyr::rename(cell = Sample)
cell_metadata_PRJNA431392 <- cell_metadata_PRJNA431392 |>
dplyr::filter(`3184` == 1) |>
dplyr::mutate(developmental_stage = droplevels(developmental_stage))matrix_readcount_PRJNA431392_pseudobulk_EPI <- purrr::map(
levels(cell_metadata_PRJNA431392$developmental_stage), \(x) {
cells_in_group <- cell_metadata_PRJNA431392 |>
dplyr::filter(
developmental_stage == x,
lineage == "EPI"
) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_PRJNA431392[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(levels(cell_metadata_PRJNA431392$developmental_stage)) |>
as.matrix()
matrix_readcount_PRJNA431392_pseudobulk_TE <- purrr::map(
levels(cell_metadata_PRJNA431392$developmental_stage), \(x) {
cells_in_group <- cell_metadata_PRJNA431392 |>
dplyr::filter(
developmental_stage == x,
lineage == "TE"
) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_PRJNA431392[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(levels(cell_metadata_PRJNA431392$developmental_stage)) |>
as.matrix()
matrix_readcount_PRJNA431392_pseudobulk_HYP <- purrr::map(
levels(cell_metadata_PRJNA431392$developmental_stage), \(x) {
cells_in_group <- cell_metadata_PRJNA431392 |>
dplyr::filter(
developmental_stage == x,
lineage == "PE"
) |>
dplyr::pull(cell)
Matrix::rowSums(matrix_readcount_PRJNA431392[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(levels(cell_metadata_PRJNA431392$developmental_stage)) |>
as.matrix()
# clean up
rm(matrix_readcount_PRJNA431392)
rm(cell_metadata_PRJNA431392)Takashima et al
Resetting transcription factor control circuitry toward ground-state pluripotency in human
- BioProject Accession: PRJEB7132
- ArrayExpress Accession: E-MTAB-2857
- Notebook: https://jlduan.github.io/replica/j.cell.2014.08.029
matrix_readcount_PRJEB7132 <- load_matrix(
file.path(PROJECT_DIR, "raw/public/PRJEB7132")
)
matrix_readcount_PRJEB7132_pseudobulk <- cbind(
H9 = Matrix::rowSums(
matrix_readcount_PRJEB7132[, c("ERS537888", "ERS537890", "ERS537878")]
),
H9_reset = Matrix::rowSums(
matrix_readcount_PRJEB7132[, c("ERS537884", "ERS537881", "ERS537876")]
)
) |>
as.matrix()
# clean up
rm(matrix_readcount_PRJEB7132)Dong et al
Derivation of trophoblast stem cells from naïve human pluripotent stem cells
- BioProject Accession: PRJNA576801
- GEO Accession: GSE138688
- Notebook: https://jlduan.github.io/replica/eLife.52504
matrix_readcount_PRJNA576801 <- load_matrix(
file.path(PROJECT_DIR, "raw/public/PRJNA576801")
)
geo_info_PRJNA576801 <- tibble::tribble(
~sample_name, ~sample_annotation, ~group, ~cell_line,
# "GSM4116149", "H9 primed TSC Bulk RNA-seq", "Primed hPSC (hTSC medium)", "H9",
# "GSM4116150", "AN primed TSC Bulk RNA-seq", "Primed hPSC (hTSC medium)", "AN",
"GSM4116151", "H9 primed Bulk RNA-seq", "Primed hPSC", "H9",
"GSM4116152", "AN primed Bulk RNA-seq", "Primed hPSC", "AN",
# "GSM4116153", "H9 primed StemPro Bulk RNA-seq", "Re-primed hPSC", "H9",
# "GSM4116154", "H9 capacitated 1 Bulk RNA-seq", "Capacitated hPSC", "H9",
# "GSM4116155", "H9 capacitated 2 Bulk RNA-seq", "Capacitated hPSC", "H9",
"GSM4116156", "H9 naïve Bulk RNA-seq", "Naïve hPSC", "H9",
"GSM4116157", "AN naïve Bulk RNA-seq", "Naïve hPSC", "AN",
"GSM4116158", "H9 naïve TSC Bulk RNA-seq", "Naïve hTSC", "H9",
"GSM4116159", "AN naïve TSC Bulk RNA-seq", "Naïve hTSC", "AN",
"GSM4116160", "WIBR3 naïve TSC Bulk RNA-seq", "Naïve hTSC", "WIBR3",
# "GSM4116161", "H9 EVT Bulk RNA-seq", "EVT", "H9",
# "GSM4116162", "AN EVT Bulk RNA-seq", "EVT", "AN",
# "GSM4116163", "H9 STB Bulk RNA-seq", "STB", "H9",
# "GSM4116164", "AN STB Bulk RNA-seq", "STB", "AN",
"GSM4276363", "H9 naïve TSC rep 2 Bulk RNA-seq", "Naïve hTSC", "H9",
"GSM4276364", "AN naïve TSC rep 2 Bulk RNA-seq", "Naïve hTSC", "AN",
# "GSM4276365", "BT5 hTSC rep 1 Bulk RNA-seq", "BT5 hTSCs", "BT5",
# "GSM4276366", "BT5 hTSC rep 2 Bulk RNA-seq", "BT5 hTSCs", "BT5"
)
cell_metadata_PRJNA576801 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw/public/PRJNA576801",
"SraRunTable.txt"
),
delim = ","
) |>
dplyr::select(
sample_name = "Sample Name",
run = "Run",
biosample = "BioSample",
source_name
) |>
dplyr::mutate(
source_name = stringr::str_remove(
string = source_name, pattern = "\\s.+$"
),
source_name = factor(
source_name,
levels = c("hPSC", "hTSC", "EVT", "STB"),
)
) |>
dplyr::left_join(geo_info_PRJNA576801) |>
dplyr::mutate(cell_line = factor(cell_line)) |>
tidyr::drop_na() |>
dplyr::mutate(group = factor(group))matrix_readcount_PRJNA576801_pseudobulk_lineage <- purrr::map(
levels(cell_metadata_PRJNA576801$group), \(x) {
cells_in_group <- cell_metadata_PRJNA576801 |>
dplyr::filter(
group == x
) |>
dplyr::pull(sample_name)
Matrix::rowSums(matrix_readcount_PRJNA576801[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(levels(cell_metadata_PRJNA576801$group)) |>
as.matrix()
colnames(matrix_readcount_PRJNA576801_pseudobulk_lineage) <-
stringr::str_replace(
string = colnames(matrix_readcount_PRJNA576801_pseudobulk_lineage),
pattern = " ", replacement = "_"
)
rm(matrix_readcount_PRJNA576801)
rm(geo_info_PRJNA576801)
rm(cell_metadata_PRJNA576801)Cinkornpumin et al
- BioProject Accession: PRJNA638350
- GEO Accession: GSE152101
- Notebook: https://jlduan.github.io/replica/j.stemcr.2020.06.003
matrix_readcount_PRJNA638350 <- load_matrix(
file.path(PROJECT_DIR, "raw/public/PRJNA638350")
)
geo_info_PRJNA638350 <- tibble::tribble(
~cell, ~sample_description, ~source_name2, ~group, ~cell_line,
"GSM4603117", "BT2 hTSC Rep1", "BT2 hTSC", "hTSC", "BT2",
"GSM4603118", "BT2 hTSC Rep2", "BT2 hTSC", "hTSC", "BT2",
"GSM4603119", "BT2 hTSC Rep3", "BT2 hTSC", "hTSC", "BT2",
"GSM4603120", "BT2 hTSC Rep4", "BT2 hTSC", "hTSC", "BT2",
"GSM4603121", "CT1 hTSCs Rep1", "CT1 hTSC", "hTSC", "CT1",
"GSM4603122", "CT1 hTSCs Rep2", "CT1 hTSC", "hTSC", "CT1",
"GSM4603123", "CT1 hTSCs Rep3", "CT1 hTSC", "hTSC", "CT1",
"GSM4603124", "CT1 hTSCs Rep4", "CT1 hTSC", "hTSC", "CT1",
"GSM4603125", "CT1 hTSCs Rep5", "CT1 hTSC", "hTSC", "CT1",
"GSM4603126", "CT1 hTSCs Rep6", "CT1 hTSC", "hTSC", "CT1",
"GSM4603127", "CT1 hTSCs Rep7", "CT1 hTSC", "hTSC", "CT1",
"GSM4603128", "CT3 hTSCs Rep1", "CT3 hTSC", "hTSC", "CT3",
"GSM4603129", "CT3 hTSCs Rep2", "CT3 hTSC", "hTSC", "CT3",
"GSM4603130", "CT3 hTSCs Rep3", "CT3 hTSC", "hTSC", "CT3",
"GSM4603131", "CT3 hTSCs Rep4", "CT3 hTSC", "hTSC", "CT3",
# "GSM4603132", "UCLA1 Primed hESCs Rep1", "UCLA1 Primed hESC", "Primed hESC", "UCLA1",
# "GSM4603133", "UCLA1 Primed hESCs Rep2", "UCLA1 Primed hESC", "Primed hESC", "UCLA1",
# "GSM4603134", "UCLA1 Primed hESCs Rep3", "UCLA1 Primed hESC", "Primed hESC", "UCLA1",
"GSM4603135", "WIBR3 Primed hESCs Rep 1", "WIBR3 Primed hESC", "Primed hESC", "WIBR3",
"GSM4603136", "WIBR3 Primed hESCs Rep 2", "WIBR3 Primed hESC", "Primed hESC", "WIBR3",
"GSM4603137", "WIBR3 Primed hESCs Rep 3", "WIBR3 Primed hESC", "Primed hESC", "WIBR3",
"GSM4603138", "WIBR3 Primed hESCs Rep 4", "WIBR3 Primed hESC", "Primed hESC", "WIBR3",
# "GSM4603139", "WIBR3 Naive hESCs Rep 1", "WIBR3 Naive hESC", "Primed hESC", "WIBR3",
# "GSM4603140", "WIBR3 Naive hESCs Rep 2", "WIBR3 Naive hESC", "Primed hESC", "WIBR3",
"GSM4603139", "WIBR3 Naive hESCs Rep 1", "WIBR3 Naive hESC", "Naive hESC", "WIBR3",
"GSM4603140", "WIBR3 Naive hESCs Rep 2", "WIBR3 Naive hESC", "Naive hESC", "WIBR3",
"GSM4603141", "WIBR3 tdhTSC Line 1 Rep 1", "WIBR3 tdhTSC", "tdhTSC", "WIBR3",
"GSM4603142", "WIBR3 tdhTSC Line 1 Rep 2", "WIBR3 tdhTSC", "tdhTSC", "WIBR3",
"GSM4603143", "WIBR3 tdhTSC Line 2 Rep1", "WIBR3 tdhTSC", "tdhTSC", "WIBR3",
"GSM4603144", "WIBR3 tdhTSC Line 2 Rep 2", "WIBR3 tdhTSC", "tdhTSC", "WIBR3",
"GSM4603145", "WIBR3 tdhTSC Line 3 Rep 1", "WIBR3 tdhTSC", "tdhTSC", "WIBR3",
"GSM4603146", "UCLA1 tdhTSC Line 1 Rep 1", "UCLA1 tdhTSC", "tdhTSC", "UCLA1",
"GSM4603147", "UCLA1 tdhTSC Line 1 Rep 2", "UCLA1 tdhTSC", "tdhTSC", "UCLA1",
"GSM4603148", "FT190", "FT190", "FT190", "FT190",
"GSM4603149", "Hec116", "Hec116", "Hec116", "Hec116"
)
cell_metadata_PRJNA638350 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw/public/PRJNA638350",
"SraRunTable.txt"
),
delim = ","
) |>
dplyr::rename_with(~ tolower(gsub(" ", "_", .x, fixed = TRUE))) |>
dplyr::select(
cell = sample_name,
run,
biosample,
source_name,
cell_type
) |>
dplyr::left_join(geo_info_PRJNA638350, by = "cell") |>
dplyr::mutate(
group = factor(
group,
levels = c("Naive hESC", "Primed hESC", "tdhTSC", "hTSC", "FT190", "Hec116")
)
) |>
tidyr::drop_na()matrix_readcount_PRJNA638350_pseudobulk_lineage <- purrr::map(
levels(cell_metadata_PRJNA638350$group), \(x) {
cells_in_group <- cell_metadata_PRJNA638350 |>
dplyr::filter(
group == x
) |>
dplyr::pull(cell)
if (length(cells_in_group) > 1) {
m <- Matrix::rowSums(matrix_readcount_PRJNA638350[, cells_in_group])
} else {
m <- matrix_readcount_PRJNA638350[, cells_in_group]
}
return(m)
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(levels(cell_metadata_PRJNA638350$group)) |>
as.matrix()
colnames(matrix_readcount_PRJNA638350_pseudobulk_lineage) <-
stringr::str_replace(
colnames(matrix_readcount_PRJNA638350_pseudobulk_lineage),
pattern = " ", replacement = "_"
)
# clean up
rm(matrix_readcount_PRJNA638350)
rm(geo_info_PRJNA638350)
rm(cell_metadata_PRJNA638350)Lv et al
- BioProject Accession: PRJNA516921
- GEO Accession: GSE125616
- Notebook: https://jlduan.github.io/replica/journal.pbio.3000187
matrix_readcount_PRJNA516921 <- load_matrix(
file.path(PROJECT_DIR, "raw/public/PRJNA516921")
)
geo_info_PRJNA516921 <- read_delim(
file = file.path(
PROJECT_DIR,
"raw/public/PRJNA516921",
"notebooks",
"sample.csv"
),
delim = ","
)
cell_metadata_PRJNA516921 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw/public/PRJNA516921",
"SraRunTable.txt"
),
delim = ","
) |>
dplyr::rename_with(~ tolower(gsub(" ", "_", .x, fixed = TRUE))) |>
dplyr::select(
cell = sample_name,
run,
biosample,
development_day,
source_name,
stage
) |>
dplyr::left_join(
geo_info_PRJNA516921 |>
select(Accession, Title),
by = c("cell" = "Accession")
) |>
dplyr::rename_all(tolower) |>
dplyr::rename(
developmental_stage = "development_day",
) |>
dplyr::mutate(
developmental_stage = stringr::str_replace(
string = developmental_stage,
pattern = "day",
replacement = "Day "
),
developmental_stage = factor(
developmental_stage,
levels = developmental_stage |>
unique() |>
str_sort(numeric = TRUE)
),
stage = stringr::str_to_title(string = stage),
stage = factor(
stage,
levels = c(
"Blastocyst", "Implantation",
"Post-Implantation", "Endometrial"
)
)
)
cell_metadata_PRJNA516921 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw/public/PRJNA516921",
"exploring/embedding_ncomponents14_seed20200317.csv"
)
) |>
dplyr::left_join(
cell_metadata_PRJNA516921,
by = c("cell" = "cell")
) |>
dplyr::select(
cell:y_umap, run:dplyr::last_col()
) |>
dplyr::filter(
!(
louvain == 3 | (
developmental_stage == "Day 6" & louvain == 1
) | (
developmental_stage %in% c("Day 13", "Day 14")
)
)
) |>
dplyr::mutate(
lineage = dplyr::case_when(
cell %in% c(
"GSM3578305",
"GSM3578308",
"GSM3578339",
"GSM3578505",
"GSM3578651",
"GSM3578653",
"GSM3578682",
"GSM3578696",
"GSM3578697",
"GSM3578707",
"GSM3578729"
) ~ "PE",
cell %in% c(
"GSM3578300",
"GSM3578302",
"GSM3578365",
"GSM3578492",
"GSM3578515",
"GSM3578666",
"GSM3578668",
"GSM3578714",
"GSM3578716",
"GSM3578728"
) ~ "EPI",
TRUE ~ "TE"
),
lineage = factor(
lineage,
levels = c("EPI", "PE", "TE")
)
)matrix_readcount_PRJNA516921_pseudobulk_TE <- purrr::map(
levels(cell_metadata_PRJNA516921$developmental_stage), \(x) {
cells_in_group <- cell_metadata_PRJNA516921 |>
dplyr::filter(
lineage == "TE",
developmental_stage == x
) |>
dplyr::pull(cell)
if (length(cells_in_group) > 1) {
m <- Matrix::rowSums(matrix_readcount_PRJNA516921[, cells_in_group])
} else {
m <- matrix_readcount_PRJNA516921[, cells_in_group]
}
return(m)
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(cell_metadata_PRJNA516921 |>
dplyr::filter(lineage == "TE") |>
dplyr::pull(developmental_stage) |>
droplevels() |>
levels()) |>
as.matrix()
rm(matrix_readcount_PRJNA516921)
rm(geo_info_PRJNA516921)
rm(cell_metadata_PRJNA516921)Linneberg-Agerholm et al
- BioProject Accession: PRJNA574150
- GEO Accession: GSE138012
- Notebook: https://jlduan.github.io/replica/dev.180620
matrix_readcount_PRJNA574150 <- load_matrix(
file.path(PROJECT_DIR, "raw/public/PRJNA574150")
)
geo_info_PRJNA574150 <- tibble::tribble(
~cell, ~sample_description,
"GSM4096610", "H9_pES_R1",
"GSM4096611", "H9_pES_R2",
"GSM4096612", "H9_t2iLGo_nES_R2",
"GSM4096613", "H9_t2iLGo_nES_R1",
"GSM4096614", "H9_t2iLGo_nES_R3",
"GSM4096615", "H9_h2iL_nES_R1",
"GSM4096616", "H9_h2iL_nES_R2",
"GSM4096617", "H9_h2iL_nES_R3",
"GSM4096618", "H9_t2iLGo_PrE_R2",
"GSM4096619", "H9_t2iLGo_PrE_R3",
"GSM4096620", "H9_t2iLGo_PrE_R1",
"GSM4096621", "H9_h2iL_PrE_R2",
"GSM4096622", "H9_h2iL_PrE_R3",
"GSM4096623", "H9_h2iL_PrE_R1",
"GSM4096624", "HUES4_pES_R1",
"GSM4096625", "HUES4_pES_R2",
"GSM4096626", "H9_ADE_R1",
"GSM4096627", "H9_ADE_R2",
"GSM4096628", "HUES4_ADE_R2",
"GSM4096629", "HUES4_ADE_R1",
"GSM4096630", "H9_RSeT_nES_R1",
"GSM4096631", "H9_RSeT_nES_R2",
"GSM4096632", "H9_t2iLGo_nEnd_R1",
"GSM4096633", "H9_t2iLGo_nEnd_R2",
"GSM4096634", "H9_t2iLGo_nEnd_R3",
"GSM4096635", "H9_RSeT_PrE_R1-1",
"GSM4096636", "H9_RSeT_PrE_R2-1",
"GSM4096637", "H9_DE_R1",
"GSM4096638", "H9_DE_R2",
"GSM4096639", "H9_DE_R3",
"GSM4096640", "H9_RSeT_PrE_R1-2",
"GSM4096641", "H9_RSeT_PrE_R2-2",
"GSM4096642", "H9_RSeT_nEnd_R1",
"GSM4096643", "H9_RSeT_nEnd_R2",
"GSM4096644", "H9_RSeT_nEnd_R3"
)
cell_metadata_PRJNA574150 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw/public/PRJNA574150",
"SraRunTable.txt"
),
delim = ","
) |>
dplyr::rename_with(~ tolower(gsub(" ", "_", .x, fixed = TRUE))) |>
dplyr::select(
cell = sample_name,
run,
biosample,
cell_type,
background,
source_name,
stage
) |>
dplyr::left_join(geo_info_PRJNA574150, by = "cell") |>
dplyr::mutate(
group = stringr::str_remove(
string = sample_description,
pattern = "_R\\d.*$"
),
lineage = stringr::str_remove(
string = group, pattern = ".+_"
),
lineage = factor(
lineage,
levels = c("nES", "pES", "PrE", "nEnd", "DE", "ADE")
)
) |>
dplyr::rename(cell_line = "background")matrix_readcount_PRJNA574150_pseudobulk_lineage <- purrr::map(
levels(cell_metadata_PRJNA574150$lineage), \(x) {
cells_in_group <- cell_metadata_PRJNA574150 |>
dplyr::filter(
lineage == x,
) |>
dplyr::pull(cell)
if (length(cells_in_group) > 1) {
m <- Matrix::rowSums(matrix_readcount_PRJNA574150[, cells_in_group])
} else {
m <- matrix_readcount_PRJNA574150[, cells_in_group]
}
return(m)
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(levels(cell_metadata_PRJNA574150$lineage)) |>
as.matrix()
rm(matrix_readcount_PRJNA574150)
rm(geo_info_PRJNA574150)
rm(cell_metadata_PRJNA574150)Principal component analysis
# prepare features
table_s3_sheet1 <- readxl::read_excel(
path = file.path(
PROJECT_DIR,
"docs",
"Reconstituting_the_transcriptome_and_DNA_methylome_landscapes_of_human_implantation",
"41586_2019_1500_MOESM3_ESM",
"Supplementary Table 3 Lineage_Markers.xlsx"
),
sheet = "Sheet1"
)
table_s3_sheet1 |>
head() |>
knitr::kable()| gene | myAUC | avg_diff | power | pct.1 | pct.2 | cluster |
|---|---|---|---|---|---|---|
| DPPA5 | 0.987 | 4.287821 | 0.974 | 0.985 | 0.247 | EPI |
| IFITM1 | 0.987 | 3.334143 | 0.974 | 0.997 | 0.593 | EPI |
| POU5F1 | 0.986 | 3.814455 | 0.972 | 0.988 | 0.355 | EPI |
| KHDC3L | 0.970 | 4.646703 | 0.940 | 0.958 | 0.123 | EPI |
| TDGF1 | 0.970 | 3.947136 | 0.940 | 0.958 | 0.202 | EPI |
| ESRG | 0.964 | 3.447912 | 0.928 | 0.961 | 0.393 | EPI |
features_hg_EPI <- rownames(matrix_readcount_use)[
stringr::str_remove(
string = rownames(matrix_readcount_use),
pattern = "^E.+_"
) %in% (table_s3_sheet1 |>
dplyr::filter(
cluster == "EPI"
) |>
dplyr::pull(gene))
] |>
unique()
features_hg_cy_EPI_hg <- features_hg_cy |>
dplyr::filter(hg %in% features_hg_EPI) |>
dplyr::pull(hg)
features_hg_cy_EPI_cy <- features_hg_cy |>
dplyr::filter(hg %in% features_hg_EPI) |>
dplyr::pull(cy)features_hg_TE <- rownames(matrix_readcount_use)[
stringr::str_remove(
string = rownames(matrix_readcount_use),
pattern = "^E.+_"
) %in% (table_s3_sheet1 |>
dplyr::filter(
cluster == "TE"
) |>
dplyr::pull(gene))
] |>
unique()
features_hg_cy_TE_hg <- features_hg_cy |>
dplyr::filter(hg %in% features_hg_TE) |>
dplyr::pull(hg)
features_hg_cy_TE_cy <- features_hg_cy |>
dplyr::filter(hg %in% features_hg_TE) |>
dplyr::pull(cy)features_hg_HYP <- rownames(matrix_readcount_use)[
stringr::str_remove(
string = rownames(matrix_readcount_use),
pattern = "^E.+_"
) %in% (table_s3_sheet1 |>
dplyr::filter(
cluster == "PE"
) |>
dplyr::pull(gene))
] |>
unique()
features_hg_cy_HYP_hg <- features_hg_cy |>
dplyr::filter(hg %in% features_hg_HYP) |>
dplyr::pull(hg)
features_hg_cy_HYP_cy <- features_hg_cy |>
dplyr::filter(hg %in% features_hg_HYP) |>
dplyr::pull(cy)EPI
# z-score
clusters_selected_blastoid <- c("2", "3", "11")
matrix_readcount_pca <- cbind(
matrix_readcount_blastoid_pseudobulk[, clusters_selected_blastoid],
#
matrix_readcount_stem_pseudobulk[, c("LW50"), drop = FALSE],
#
matrix_readcount_PRJEB11202_pseudobulk_EPI,
matrix_readcount_PRJNA562548_pseudobulk_EPI,
matrix_readcount_PRJNA431392_pseudobulk_EPI,
matrix_readcount_PRJEB7132_pseudobulk,
matrix_readcount_PRJNA576801_pseudobulk_lineage[
, c("Naïve_hPSC", "Primed_hPSC")
],
matrix_readcount_PRJNA638350_pseudobulk_lineage[
, c("Naive_hESC", "Primed_hESC")
]
)
colnames(matrix_readcount_pca) <- paste(
rep(
c(
"Blastoid",
#
"Blastoid",
#
"PRJEB11202",
"PRJNA562548",
"PRJNA431392",
"PRJEB7132",
"PRJNA576801",
"PRJNA638350"
),
times = c(
length(clusters_selected_blastoid),
#
ncol(matrix_readcount_stem_pseudobulk[, c("LW50"), drop = FALSE]),
#
ncol(matrix_readcount_PRJEB11202_pseudobulk_EPI),
ncol(matrix_readcount_PRJNA562548_pseudobulk_EPI),
ncol(matrix_readcount_PRJNA431392_pseudobulk_EPI),
ncol(matrix_readcount_PRJEB7132_pseudobulk),
length(c("Naïve_hPSC", "Primed_hPSC")),
length(c("Naive_hESC", "Primed_hESC"))
)
),
colnames(matrix_readcount_pca),
sep = ":"
)pca_out_EPI <- perform_pca(
matrix = matrix_readcount_pca,
features = features_hg_cy_EPI_hg,
min_counts = 1
)
embedding_pca_EPI <- pca_out_EPI$x |>
as.data.frame() |>
tibble::rownames_to_column(var = "sample_name") |>
dplyr::select(sample_name:PC3) |>
dplyr::rename(
x_pca = PC1,
y_pca = PC2,
z_pca = PC3
) |>
dplyr::relocate(x_pca, y_pca, z_pca, .after = dplyr::last_col()) |>
dplyr::mutate(
lineage = stringr::str_remove(string = sample_name, pattern = "^P.+:"),
dataset = stringr::str_remove(string = sample_name, pattern = ":.+$")
)TE
clusters_selected_blastoid <- c("0", "1", "8", "9", "12", "17")
matrix_readcount_pca <- cbind(
matrix_readcount_blastoid_pseudobulk[, clusters_selected_blastoid],
#
matrix_readcount_stem_pseudobulk[, c("LW52"), drop = FALSE],
#
matrix_readcount_PRJEB11202_pseudobulk_TE,
matrix_readcount_PRJNA431392_pseudobulk_TE,
#
matrix_readcount_PRJNA576801_pseudobulk_lineage,
matrix_readcount_PRJNA638350_pseudobulk_lineage[
,
c("Naive_hESC", "Primed_hESC", "tdhTSC", "hTSC")
],
matrix_readcount_PRJNA516921_pseudobulk_TE
)
# name columns
colnames(matrix_readcount_pca) <- paste(
rep(
c(
"Blastoid",
#
"Blastoid",
#
"PRJEB11202",
"PRJNA431392",
#
"PRJNA576801",
"PRJNA638350",
"PRJNA516921"
),
times = c(
ncol(matrix_readcount_blastoid_pseudobulk[
,
clusters_selected_blastoid
]),
#
ncol(matrix_readcount_stem_pseudobulk[, c("LW52"), drop = FALSE]),
#
ncol(matrix_readcount_PRJEB11202_pseudobulk_TE),
ncol(matrix_readcount_PRJNA431392_pseudobulk_TE),
#
ncol(matrix_readcount_PRJNA576801_pseudobulk_lineage),
length(c("Naive_hESC", "Primed_hESC", "tdhTSC", "hTSC")),
ncol(matrix_readcount_PRJNA516921_pseudobulk_TE)
)
),
colnames(matrix_readcount_pca),
sep = ":"
)
colnames(matrix_readcount_pca) <- stringr::str_replace_all(
string = colnames(matrix_readcount_pca),
pattern = " ",
replacement = "_"
)
matrix_readcount_pca <- matrix_readcount_pca[
, colnames(matrix_readcount_pca) %in% c(
"Blastoid:0",
"Blastoid:1",
"Blastoid:8",
"Blastoid:9",
"Blastoid:12",
"Blastoid:17",
"Blastoid:LW52",
"PRJEB11202:E5",
"PRJEB11202:E6",
"PRJEB11202:E7",
"PRJNA431392:E6",
"PRJNA431392:E8",
"PRJNA431392:E10",
"PRJNA431392:E12",
"PRJNA576801:Naïve_hTSC",
"PRJNA638350:tdhTSC",
"PRJNA638350:hTSC",
"PRJNA516921:Day_6",
"PRJNA516921:Day_7",
"PRJNA516921:Day_8",
"PRJNA516921:Day_9",
"PRJNA516921:Day_10"
)
]pca_out_TE <- perform_pca(
matrix = matrix_readcount_pca,
features = features_hg_cy_TE_hg,
min_counts = 1
)
embedding_pca_TE <- pca_out_TE$x |>
as.data.frame() |>
tibble::rownames_to_column(var = "sample_name") |>
dplyr::select(sample_name:PC3) |>
dplyr::rename(
x_pca = PC1,
y_pca = PC2,
z_pca = PC3
) |>
dplyr::relocate(x_pca, y_pca, z_pca, .after = dplyr::last_col()) |>
dplyr::mutate(
lineage = stringr::str_remove(string = sample_name, pattern = "^P.+:"),
dataset = stringr::str_remove(string = sample_name, pattern = ":.+$")
)HYP
clusters_selected_blastoid <- c("18", "15", "16", "13")
matrix_readcount_pca <- cbind(
matrix_readcount_blastoid_pseudobulk[
,
clusters_selected_blastoid,
drop = FALSE
],
#
matrix_readcount_stem_pseudobulk[, c("LW51"), drop = FALSE],
#
matrix_readcount_PRJEB11202_pseudobulk_HYP,
matrix_readcount_PRJNA562548_pseudobulk_HYP,
matrix_readcount_PRJNA431392_pseudobulk_HYP,
#
matrix_readcount_PRJNA574150_pseudobulk_lineage[, "nEnd", drop = FALSE]
)
# name columns
colnames(matrix_readcount_pca) <- paste(
rep(
c(
"Blastoid",
#
"Blastoid",
#
"PRJEB11202",
"PRJNA562548",
"PRJNA431392",
#
"PRJNA574150"
),
times = c(
length(clusters_selected_blastoid),
#
ncol(matrix_readcount_stem_pseudobulk[, c("LW51"), drop = FALSE]),
#
ncol(matrix_readcount_PRJEB11202_pseudobulk_HYP),
ncol(matrix_readcount_PRJNA562548_pseudobulk_HYP),
ncol(matrix_readcount_PRJNA431392_pseudobulk_HYP),
#
length("nEnd")
)
),
colnames(matrix_readcount_pca),
sep = ":"
)
colnames(matrix_readcount_pca) <- stringr::str_replace_all(
string = colnames(matrix_readcount_pca),
pattern = " ",
replacement = "_"
)pca_out_HYP <- perform_pca(
matrix = matrix_readcount_pca,
features = features_hg_cy_HYP_hg,
min_counts = 1
)
embedding_pca_HYP <- pca_out_HYP$x |>
as.data.frame() |>
tibble::rownames_to_column(var = "sample_name") |>
dplyr::select(sample_name:PC3) |>
dplyr::rename(
x_pca = PC1,
y_pca = PC2,
z_pca = PC3
) |>
dplyr::relocate(x_pca, y_pca, z_pca, .after = dplyr::last_col()) |>
dplyr::mutate(
lineage = stringr::str_remove(string = sample_name, pattern = "^P.+:"),
dataset = stringr::str_remove(string = sample_name, pattern = ":.+$")
)Visualization, joint
# assign colors
studies <- tibble::tribble(
~bioproject, ~description,
"Blastoid", "Blastoid",
"PRJEB11202", "Petropoulos et al., 2016",
"PRJEB7132", "Takashima et al., 2014",
"PRJNA431392", "Zhou et al., 2019",
"PRJNA516921", "lv et al., 2019",
"PRJNA562548", "Xiang et al., 2020",
"PRJNA574150", "Linneberg-Agerholm et al., 2019",
"PRJNA576801", "Dong et al., 2020",
"PRJNA638350", "Cinkornpumin et al., 2020"
) |>
mutate(
color = c(
"#F8766D",
"#D39200",
"#93AA00",
"#00BA38",
"#00C19F",
"#00B9E3",
"#619CFF",
"#DB72FB",
"#FF61C3"
)
)embedding_pca_EPI <- embedding_pca_EPI |>
dplyr::mutate(dataset = factor(dataset, levels = studies$bioproject))
embedding_pca_HYP <- embedding_pca_HYP |>
dplyr::mutate(
y_pca = 0 - y_pca,
dataset = factor(dataset, levels = studies$bioproject),
)
embedding_pca_TE <- embedding_pca_TE |>
dplyr::mutate(
dataset = factor(dataset, levels = studies$bioproject),
lineage = stringr::str_replace(
string = lineage, pattern = "Day_", replacement = "E"
)
)# compose
p1 <- plot_pca_embedding(
data = embedding_pca_EPI,
x = x_pca,
y = y_pca,
color = dataset,
title = "EPI; PCA",
legend_x = 0.5,
legend_y = 0.995,
min_segment_length = 0
) +
add_xy_label_pca(pca_out_EPI) +
ggplot2::scale_color_manual(
values = studies$color,
breaks = studies$bioproject,
labels = studies$description,
drop = FALSE
) +
ggplot2::theme(legend.position = "bottom") +
ggplot2::guides(
colour = ggplot2::guide_legend(
override.aes = list(size = 2.5), ncol = 2,
)
)
ab <- coef(
MASS::rlm(y_pca ~ x_pca,
data = embedding_pca_EPI |>
dplyr::filter(dataset != "Blastoid")
)
)
p1 <- p1 + ggplot2::geom_abline(
intercept = ab[1], slope = ab[2], color = "#8ace7e", size = 0.2
)Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0. Please use
`linewidth` instead.
ℹ The deprecated feature was likely used in the ggplot2 package.
Please report the issue at <https://github.com/tidyverse/ggplot2/issues>.p2 <- plot_pca_embedding(
data = embedding_pca_HYP,
x = y_pca,
y = x_pca,
color = dataset,
title = "HYP; PCA",
legend_x = 0.5,
legend_y = 0.995,
min_segment_length = 0
) +
add_xy_label_pca(pca_out_HYP, "PC2", "PC1") +
ggplot2::scale_color_manual(
values = studies$color,
breaks = studies$bioproject,
labels = studies$description,
drop = FALSE
) +
ggplot2::theme(legend.position = "bottom") +
ggplot2::guides(
colour = ggplot2::guide_legend(
override.aes = list(size = 2.5), ncol = 2
)
)
ab <- coef(
MASS::rlm(x_pca ~ y_pca,
data = embedding_pca_HYP |>
dplyr::filter(dataset != "Blastoid")
)
)
p2 <- p2 + ggplot2::geom_abline(
intercept = ab[1], slope = ab[2],
color = "#ff684c", linewidth = 0.2
)
p3 <- plot_pca_embedding(
data = embedding_pca_TE,
x = x_pca,
y = y_pca,
color = dataset,
title = "TE; PCA",
legend_x = 0.5,
legend_y = 0.995,
min_segment_length = 0
) +
add_xy_label_pca(pca_out_TE) +
ggplot2::scale_color_manual(
values = studies$color,
breaks = studies$bioproject,
labels = studies$description,
drop = FALSE
) +
ggplot2::theme(legend.position = "bottom") +
ggplot2::guides(
colour = ggplot2::guide_legend(
override.aes = list(size = 2.5), ncol = 2
)
)
ab <- coef(
MASS::rlm(y_pca ~ x_pca,
data = embedding_pca_TE |>
dplyr::filter(dataset != "Blastoid")
)
)
p3 <- p3 + ggplot2::geom_abline(
intercept = ab[1], slope = ab[2],
color = "#9467bd", size = 0.2
)list(p1 + p2 + p3) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = 1, guides = "collect") +
patchwork::plot_annotation(theme = theme(plot.margin = margin())) &
theme(legend.position = "bottom", legend.direction = "horizontal")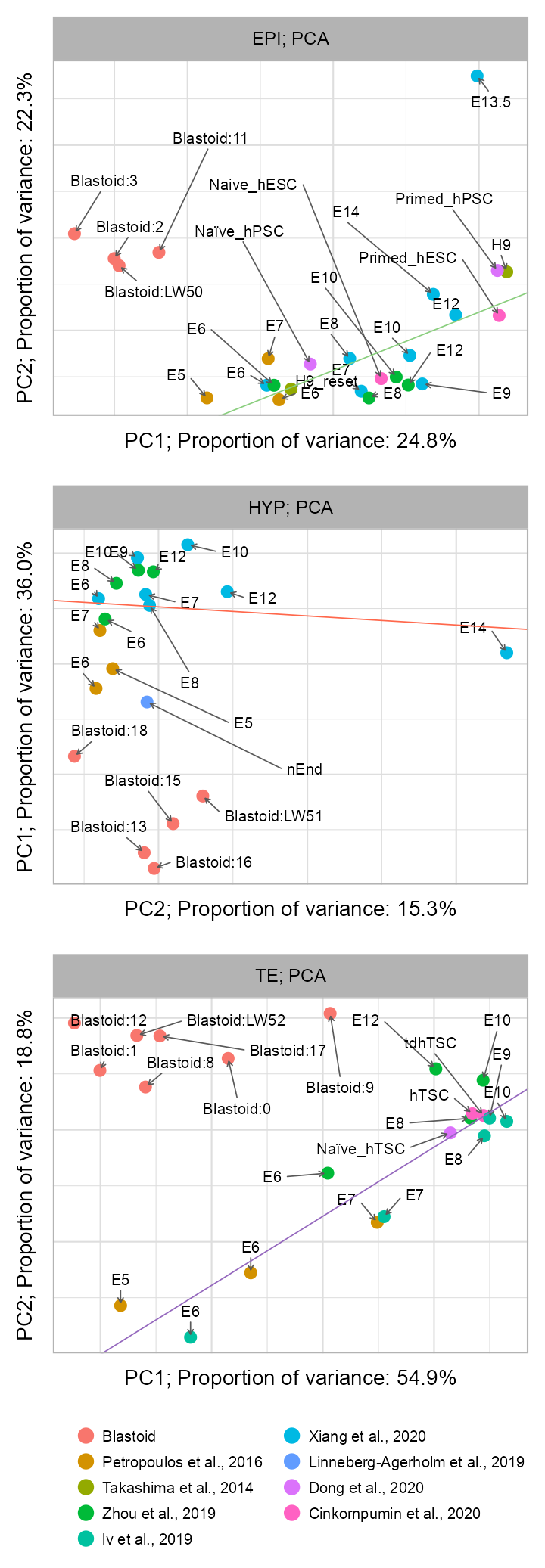
Comparison w/ cynomolgus monkeys
A developmental coordinate of pluripotency among mice, monkeys and humans
- BioProject Accession: PRJNA301445
- GEO Accession: GSE74767
- Notebook: https://jlduan.github.io/replica/nature19096
Data loading
color_palette_cluster <- c(
"0" = "#8DD3C7",
"1" = "#9EDAE5FF",
"2" = "#BEBADA",
"3" = "#FB8072",
"4" = "#80B1D3",
"5" = "#FDB462",
"6" = "#B3DE69",
"7" = "#FCCDE5",
"8" = "#DC863B",
"9" = "#BC80BD",
"10" = "#11c638",
"11" = "#BCBD22FF",
"12" = "#17BECFFF",
"13" = "#AEC7E8FF",
"14" = "#EAD3C6",
"15" = "#98DF8AFF",
"16" = "#FF9896FF",
"17" = "#C5B0D5FF",
"18" = "#C49C94FF",
"19" = "#F7B6D2FF",
"20" = "#D33F6A",
"21" = "#8E063B",
"22" = "#023FA5"
)
# prepare matrix
matrix_cpm_PRJNA301445_cy <- load_matrix(
file.path(PROJECT_DIR, "raw/public/PRJNA301445/preprocessed/cy")
)
embedding_PRJNA301445_cy <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw/public/PRJNA301445/Comparison/cy_EPI/embedding_cy.csv"
)
) |>
dplyr::mutate(
group = factor(
group,
levels = c(
"ICM",
"Pre-EPI",
"Hypoblast",
"PreE-TE",
"PreL-TE",
"PostE-EPI",
"PostL-EPI",
"Gast1",
"Gast2a",
"Gast2b",
"Post-paTE",
"VE/YE",
"EXMC",
"cyESCFF",
"cyESCoF"
# "E5.5EPI",
# "E5.5VE",
# "E6.5EPI-T hi",
# "E6.5EPI-T lo",
# "E6.5VE",
# "EpiLC",
# "2i+L ESC"
)
)
)matrix_cpm_PRJNA301445_cy_pseudobulk_lineage <- purrr::map(
levels(embedding_PRJNA301445_cy$group), \(x) {
cells_in_group <- embedding_PRJNA301445_cy |>
dplyr::filter(
group == x
) |>
dplyr::pull(cell)
Matrix::rowMeans(matrix_cpm_PRJNA301445_cy[, cells_in_group])
}
) |>
purrr::reduce(cbind) |>
`colnames<-`(levels(embedding_PRJNA301445_cy$group)) |>
as.matrix()
rm(matrix_cpm_PRJNA301445_cy)
rm(embedding_PRJNA301445_cy)# prepare features
features_hg_cy_EPI_HYP_TE_hg <- features_hg_cy |>
dplyr::filter(
hg %in% (
c(
features_hg_cy_EPI_hg,
features_hg_cy_HYP_hg,
features_hg_cy_TE_hg
) |> unique()
)
) |>
dplyr::pull(hg)
features_hg_cy_EPI_HYP_TE_cy <- features_hg_cy |>
dplyr::filter(
hg %in% (
c(
features_hg_cy_EPI_hg,
features_hg_cy_HYP_hg,
features_hg_cy_TE_hg
) |> unique()
)
) |>
dplyr::pull(cy)Correlation heatmap
matrix_corr <- cbind(
calc_cpm(
matrix_readcount_blastoid_pseudobulk
)[features_hg_cy_EPI_HYP_TE_hg, ],
matrix_cpm_PRJNA301445_cy_pseudobulk_lineage[
features_hg_cy_EPI_HYP_TE_cy,
]
)
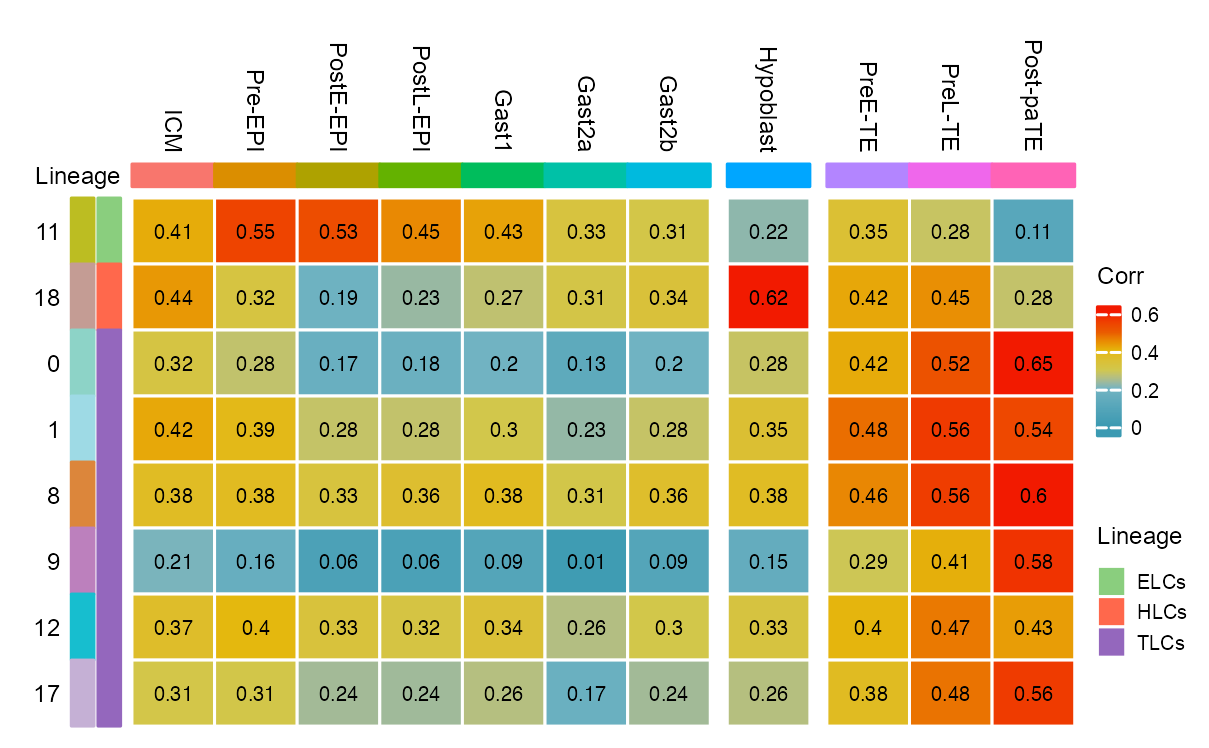
matrix_heatmap_corr_EPI_HYP_TE <- cor(log10(matrix_corr + 1) |> as.matrix())
matrix_heatmap_corr_EPI_HYP_TE <- matrix_heatmap_corr_EPI_HYP_TE[
c("11", "18", "0", "1", "8", "9", "12", "17"),
c(
"ICM", "Pre-EPI", "PostE-EPI", "PostL-EPI",
"Gast1", "Gast2a", "Gast2b",
"Hypoblast", "PreE-TE", "PreL-TE", "Post-paTE"
)
]Preview correlations.
Code
matrix_heatmap_corr_EPI_HYP_TE |>
as.data.frame() |>
tibble::rownames_to_column(var = "cluster") |>
gt::gt() |>
gt::fmt_number(columns = c(`EPI:ICM`:`TE:Post-paTE`), decimals = 2) |>
gt::data_color(
columns = c(`EPI:ICM`:`TE:Post-paTE`),
colors = scales::col_numeric(
c("#63BE7B", "#FFEB84", "#F87274"),
domain = range(matrix_heatmap_corr_EPI_HYP_TE)
)
) |>
gt::cols_align(
columns = dplyr::everything(),
align = c("center")
) |>
gt::cols_width(
dplyr::everything() ~ gt::px(60)
)| cluster | EPI:ICM | EPI:Pre-EPI | EPI:PostE-EPI | EPI:PostL-EPI | EPI:Gast1 | EPI:Gast2a | EPI:Gast2b | HYP:Hypoblast | TE:PreE-TE | TE:PreL-TE | TE:Post-paTE |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 11 | 0.41 | 0.55 | 0.53 | 0.45 | 0.43 | 0.33 | 0.31 | 0.22 | 0.35 | 0.28 | 0.11 |
| 18 | 0.44 | 0.32 | 0.19 | 0.23 | 0.27 | 0.31 | 0.34 | 0.62 | 0.42 | 0.45 | 0.28 |
| 0 | 0.32 | 0.28 | 0.17 | 0.18 | 0.20 | 0.13 | 0.20 | 0.28 | 0.42 | 0.52 | 0.65 |
| 1 | 0.42 | 0.39 | 0.28 | 0.28 | 0.30 | 0.23 | 0.28 | 0.35 | 0.48 | 0.56 | 0.54 |
| 8 | 0.38 | 0.38 | 0.33 | 0.36 | 0.38 | 0.31 | 0.36 | 0.38 | 0.46 | 0.56 | 0.60 |
| 9 | 0.21 | 0.16 | 0.06 | 0.06 | 0.09 | 0.01 | 0.09 | 0.15 | 0.29 | 0.41 | 0.58 |
| 12 | 0.37 | 0.40 | 0.33 | 0.32 | 0.34 | 0.26 | 0.30 | 0.33 | 0.40 | 0.47 | 0.43 |
| 17 | 0.31 | 0.31 | 0.24 | 0.24 | 0.26 | 0.17 | 0.24 | 0.26 | 0.38 | 0.48 | 0.56 |
ha_group <- stringr::str_remove(
string = colnames(matrix_heatmap_corr_EPI_HYP_TE),
pattern = ":.+$"
)
ha_column <- ComplexHeatmap::HeatmapAnnotation(
lineage = ComplexHeatmap::anno_simple(
colnames(matrix_heatmap_corr_EPI_HYP_TE),
# pch = anno_labels_cluster,
col = setNames(
object = scales::hue_pal()(n = ncol(matrix_heatmap_corr_EPI_HYP_TE)),
nm = colnames(matrix_heatmap_corr_EPI_HYP_TE)
),
which = "column",
pt_size = unit(2, "mm"),
pt_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 5
),
simple_anno_size = unit(2, "mm")
),
show_annotation_name = TRUE,
annotation_label = c(
"Lineage"
),
annotation_name_gp = grid::gpar(fontfamily = "Arial", fontsize = 6),
annotation_name_side = "left"
)
ha_left <- ComplexHeatmap::HeatmapAnnotation(
cluster = ComplexHeatmap::anno_simple(
rownames(matrix_heatmap_corr_EPI_HYP_TE),
col = color_palette_cluster,
which = "row",
# pt_size = unit(2, "mm"),
pt_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 5
),
simple_anno_size = unit(2, "mm")
),
#
lineage = ComplexHeatmap::anno_simple(
rownames(matrix_heatmap_corr_EPI_HYP_TE) |>
tibble::enframe() |>
dplyr::mutate(
color = case_when(
value %in% c(11) ~ "EPI",
value %in% c(18) ~ "HYP",
value %in% c(0, 1, 8, 9, 12, 17) ~ "TE",
value %in% c(10, 14) ~ "Pre-lineage",
TRUE ~ "Blastoid"
)
) |>
dplyr::pull(color),
col = setNames(
object = c("grey70", "#8ace7e", "#ff684c", "#9467bd", "#ffd8b1"),
nm = c("Blastoid", "EPI", "HYP", "TE", "Pre-lineage")
),
which = "row",
# pt_size = unit(2, "mm"),
pt_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 5
),
simple_anno_size = unit(2, "mm")
),
# ,
which = "row",
show_annotation_name = FALSE,
annotation_label = c(
"Cluster",
"Lineage"
),
annotation_name_gp = grid::gpar(fontfamily = "Arial", fontsize = 6)
)
colors_ <- as.character(
wesanderson::wes_palette("Zissou1", 10, type = "continuous")
)
col_fun <- circlize::colorRamp2(
seq(0, 0.6, 0.1),
# viridis::plasma(11)
# wesanderson::wes_palette("Zissou1", 51, type = "continuous")
colors_[c(1, 2, 3, 5, 7, 9, 10)]
)
ht <- ComplexHeatmap::Heatmap(
matrix_heatmap_corr_EPI_HYP_TE,
# name = "heatmap",
# col = wesanderson::wes_palette("Zissou1", 50, type = "continuous"),
col = col_fun,
border = NA,
rect_gp = grid::gpar(col = "white", lwd = 1),
cell_fun = function(j, i, x, y, width, height, fill) {
# grid::grid.text(
# sprintf("%.2f", corr_heatmap[i, j]), x, y,
# gp = grid::gpar(fontsize = 6)
# )
grid::grid.text(
round(matrix_heatmap_corr_EPI_HYP_TE[i, j], 2),
x, y,
gp = grid::gpar(fontsize = 5)
)
},
#
cluster_rows = FALSE,
cluster_columns = FALSE,
#
# row_labels = NA,
row_names_side = c("left"),
row_names_gp = grid::gpar(fontfamily = "Arial", fontsize = 6),
show_row_names = TRUE,
#
column_labels = stringr::str_remove(
string = colnames(matrix_heatmap_corr_EPI_HYP_TE), pattern = "^.+:"
),
column_names_side = c("top"),
column_names_gp = grid::gpar(fontfamily = "Arial", fontsize = 6),
column_names_rot = -90,
show_column_names = TRUE,
#
show_heatmap_legend = FALSE,
#
show_row_dend = FALSE,
show_column_dend = FALSE,
#
top_annotation = ha_column,
left_annotation = ha_left,
#
heatmap_legend_param = list(
title = "Corr",
title_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 6
),
legend_direction = "vertical",
labels_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 5
),
legend_height = unit(15, "mm"),
legend_width = unit(5, "mm")
),
#
column_split = ha_group,
column_gap = unit(1.5, "mm"),
column_title_gp = grid::gpar(fontfamily = "Arial", fontsize = 6),
column_title_rot = -90,
column_title = NULL
)# legend
lgd_species <- ComplexHeatmap::Legend(
title = "Species",
labels = c("Monkey", "Human"),
#
grid_height = unit(2.5, "mm"), grid_width = unit(2.5, "mm"),
#
legend_gp = grid::gpar(
fill = c("Cynomolgus Monkey" = "#3D79F3FF", "Human" = "#34A74BFF")
),
labels_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 5
),
title_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 6
)
)
lgd_colorbar <- ComplexHeatmap::Legend(
col_fun = col_fun,
title = "Corr",
grid_height = unit(1, "mm"),
grid_width = unit(2, "mm"),
legend_height = unit(10, "mm"),
legend_width = unit(2, "mm"),
title_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 6
),
labels_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 5
)
)
lgd_lineage <- ComplexHeatmap::Legend(
title = "Lineage",
labels = c("ELCs", "HLCs", "TLCs"),
#
grid_height = unit(2.5, "mm"), grid_width = unit(2.5, "mm"),
#
legend_gp = grid::gpar(
fill = c(
"ELCs" = "#8ace7e",
"HLCs" = "#ff684c",
"TLCs" = "#9467bd"
)
),
labels_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 5
),
title_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 6
)
)
pd <- ComplexHeatmap::packLegend(
# lgd_cluster,
lgd_colorbar,
lgd_lineage,
# lgd_species,
gap = unit(8, "mm"),
direction = "vertical"
)
Derived stem cells
Data loading
matrix_readcount_5iLA <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw/naive_human_PSC",
"matrix",
"xaa_Aligned.sortedByCoord.out_deduped_q10_gene_id_featureCounts.txt.gz"
),
delim = "\t",
col_names = TRUE,
skip = 1
) |>
dplyr::select(-c(2:6))
matrix_readcount_5iLA <- Matrix(
data = as.matrix(matrix_readcount_5iLA[, -1]),
dimnames = list(
matrix_readcount_5iLA[, 1, drop = TRUE],
c("5iLA-1", "5iLA-2")
),
sparse = TRUE
)Correlation heatmap
matrix_corr <- cbind(
matrix_readcount_stem_pseudobulk[, c("LW49", "LW50", "LW51", "LW52")],
"5iLA" = Matrix::rowSums(matrix_readcount_5iLA),
matrix_readcount_PRJEB7132_pseudobulk,
matrix_readcount_PRJNA576801_pseudobulk_lineage,
matrix_readcount_PRJNA638350_pseudobulk_lineage[
,
!colnames(matrix_readcount_PRJNA638350_pseudobulk_lineage) %in% c(
"FT190", "Hec116"
)
],
matrix_readcount_PRJNA574150_pseudobulk_lineage[, "nEnd", drop = FALSE]
)
colnames(matrix_corr) <- c(
"WIBR3-5i/L/A, scRNA-Seq",
"Blastoid naïve ES cells",
"Blastoid nEND",
"Blastoid TSCs",
#
"WIBR3-5i/L/A, bulk",
#
"PRJEB7132:H9 Primed",
"PRJEB7132:H9 Reset",
#
"PRJNA576801:Naïve ES cells",
"PRJNA576801:TSCs",
"PRJNA576801:Primed ES cells",
#
"PRJNA638350:Naïve ES cells",
"PRJNA638350:Primed ES cells",
"PRJNA638350:TSCs (TD)",
"PRJNA638350:TSCs",
#
"PRJNA574150:nEND"
)
features_ <- c(
features_hg_cy_EPI_hg,
features_hg_cy_HYP_hg,
features_hg_cy_TE_hg
) |> unique()column_labels <- colnames(matrix_heatmap_corr) |>
tibble::enframe(value = "sample_name") |>
dplyr::mutate(
a = stringr::str_remove(string = sample_name, pattern = ":.+$")
) |>
dplyr::left_join(
studies,
by = c("a" = "bioproject")
) |>
dplyr::select(sample_name, a, description) |>
dplyr::mutate(
label = dplyr::case_when(
is.na(description) ~ sample_name,
TRUE ~ paste(sample_name, description, sep = "; ")
),
label = stringr::str_remove(
string = label,
pattern = ".+:"
)
) |>
dplyr::pull(label)col_fun <- circlize::colorRamp2(
seq(0, 1, 0.2),
# viridis::plasma(11)
# wesanderson::wes_palette("Zissou1", 51, type = "continuous")
#
as.character(
wesanderson::wes_palette("Zissou1", 10, type = "continuous")
)[c(1, 3, 5, 7, 9, 10)]
)
ht <- ComplexHeatmap::Heatmap(
matrix_heatmap_corr,
# name = "heatmap",
# col = wesanderson::wes_palette("Zissou1", 50, type = "continuous"),
col = col_fun,
border = NA,
rect_gp = grid::gpar(col = "white", lwd = 1),
cell_fun = function(j, i, x, y, width, height, fill) {
grid::grid.text(
round(matrix_heatmap_corr[i, j], 2),
x, y,
gp = grid::gpar(fontsize = 5)
)
},
#
cluster_rows = FALSE,
cluster_columns = FALSE,
#
row_labels = column_labels,
row_names_side = c("left"),
row_names_gp = grid::gpar(fontfamily = "Arial", fontsize = 6),
show_row_names = TRUE,
#
column_labels = column_labels,
column_names_side = c("top"),
column_names_gp = grid::gpar(fontfamily = "Arial", fontsize = 6),
# column_names_rot = -90,
column_names_rot = -45,
show_column_names = TRUE,
#
show_heatmap_legend = FALSE,
#
show_row_dend = FALSE,
show_column_dend = FALSE,
#
# top_annotation = ha_column,
# left_annotation = ha_left,
#
heatmap_legend_param = list(
title = "Corr",
title_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 6
),
legend_direction = "vertical",
labels_gp = grid::gpar(
fontfamily = "Arial",
fontsize = 5
),
legend_height = unit(15, "mm"),
legend_width = unit(5, "mm")
),
#
# column_split = ha_group,
column_gap = unit(1.5, "mm"),
column_title_gp = grid::gpar(fontfamily = "Arial", fontsize = 6),
column_title_rot = -90,
column_title = NULL
)ComplexHeatmap::draw(ht)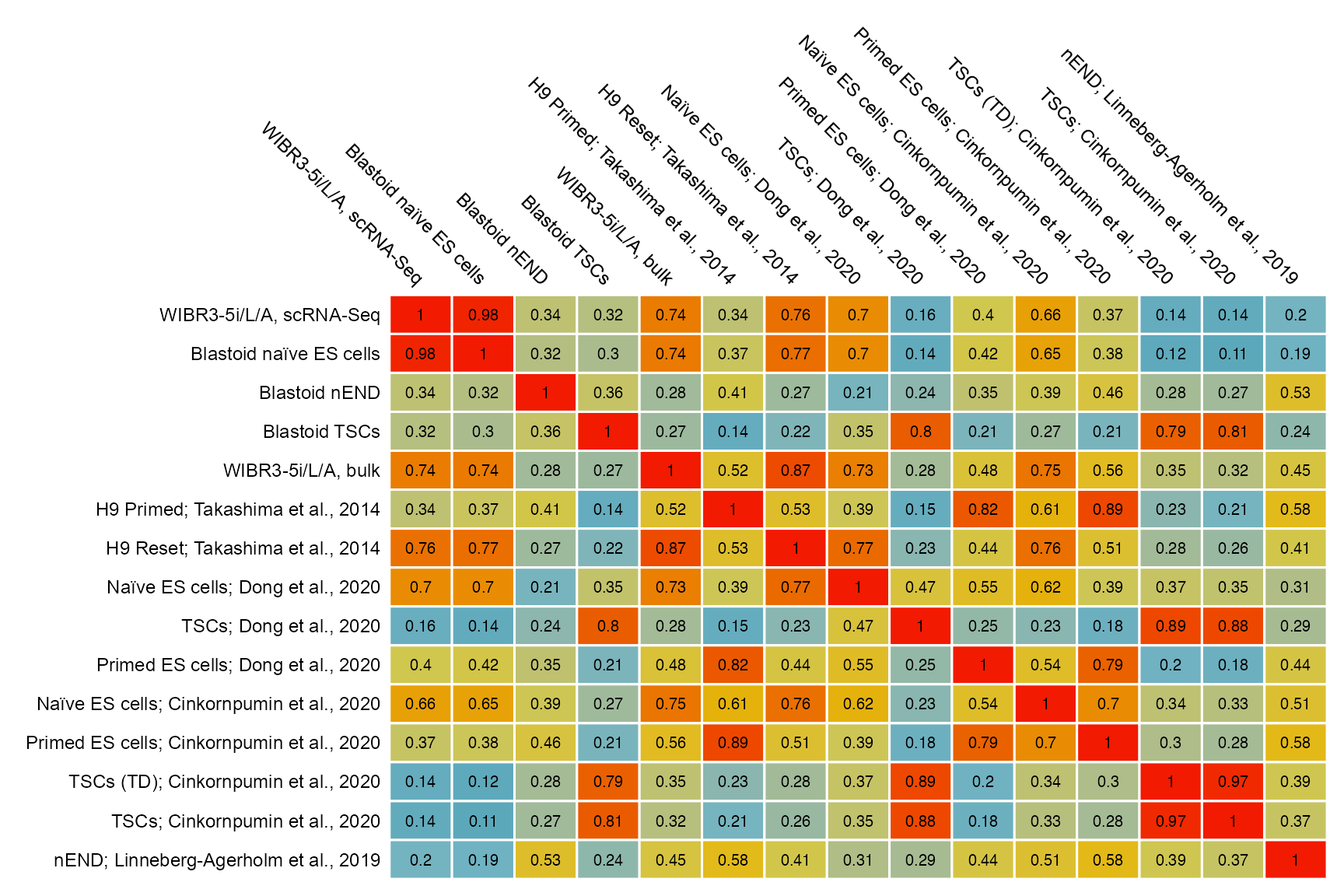
R session info
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.2.1 (2022-06-23)
os macOS Monterey 12.6
system aarch64, darwin21.6.0
ui unknown
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz America/Chicago
date 2022-09-25
pandoc 2.19.2 @ /opt/homebrew/bin/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
BiocGenerics 0.42.0 2022-04-26 [1] Bioconductor
bit 4.0.4 2020-08-04 [1] CRAN (R 4.2.0)
bit64 4.0.5 2020-08-30 [1] CRAN (R 4.2.0)
cachem 1.0.6 2021-08-19 [1] CRAN (R 4.2.0)
Cairo 1.6-0 2022-07-05 [1] CRAN (R 4.2.1)
callr 3.7.2 2022-08-22 [1] CRAN (R 4.2.1)
cellranger 1.1.0 2016-07-27 [1] CRAN (R 4.2.0)
circlize 0.4.15 2022-05-10 [1] CRAN (R 4.2.0)
cli 3.4.1 2022-09-23 [1] CRAN (R 4.2.1)
clue 0.3-61 2022-05-30 [1] CRAN (R 4.2.0)
cluster 2.1.4 2022-08-22 [2] CRAN (R 4.2.1)
codetools 0.2-18 2020-11-04 [2] CRAN (R 4.2.1)
colorspace 2.0-3 2022-02-21 [1] CRAN (R 4.2.0)
ComplexHeatmap 2.12.1 2022-08-09 [1] Bioconductor
crayon 1.5.1 2022-03-26 [1] CRAN (R 4.2.0)
devtools 2.4.4.9000 2022-09-23 [1] Github (r-lib/devtools@9e2793a)
digest 0.6.29 2021-12-01 [1] CRAN (R 4.2.0)
doParallel 1.0.17 2022-02-07 [1] CRAN (R 4.2.0)
dplyr * 1.0.99.9000 2022-09-23 [1] Github (tidyverse/dplyr@19c2be3)
ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.2.0)
evaluate 0.16 2022-08-09 [1] CRAN (R 4.2.1)
extrafont * 0.18 2022-04-12 [1] CRAN (R 4.2.0)
extrafontdb 1.0 2012-06-11 [1] CRAN (R 4.2.0)
fansi 1.0.3 2022-03-24 [1] CRAN (R 4.2.0)
farver 2.1.1 2022-07-06 [1] CRAN (R 4.2.1)
fastmap 1.1.0 2021-01-25 [1] CRAN (R 4.2.0)
forcats * 0.5.2.9000 2022-08-20 [1] Github (tidyverse/forcats@bd319e0)
foreach 1.5.2 2022-02-02 [1] CRAN (R 4.2.0)
fs 1.5.2.9000 2022-08-24 [1] Github (r-lib/fs@238032f)
generics 0.1.3 2022-07-05 [1] CRAN (R 4.2.1)
GetoptLong 1.0.5 2020-12-15 [1] CRAN (R 4.2.0)
ggplot2 * 3.3.6.9000 2022-09-12 [1] Github (tidyverse/ggplot2@a58b48c)
ggrepel 0.9.1 2021-01-15 [1] CRAN (R 4.2.0)
GlobalOptions 0.1.2 2020-06-10 [1] CRAN (R 4.2.0)
glue 1.6.2.9000 2022-04-22 [1] Github (tidyverse/glue@d47d6c7)
gt 0.7.0.9000 2022-09-23 [1] Github (rstudio/gt@4030fb7)
gtable 0.3.1.9000 2022-09-01 [1] Github (r-lib/gtable@c1a7a81)
highr 0.9 2021-04-16 [1] CRAN (R 4.2.0)
hms 1.1.2 2022-08-19 [1] CRAN (R 4.2.1)
htmltools 0.5.3 2022-07-18 [1] CRAN (R 4.2.1)
htmlwidgets 1.5.4 2022-08-23 [1] Github (ramnathv/htmlwidgets@400cf1a)
httpuv 1.6.6 2022-09-08 [1] CRAN (R 4.2.1)
IRanges 2.30.1 2022-08-18 [1] Bioconductor
iterators 1.0.14 2022-02-05 [1] CRAN (R 4.2.0)
jsonlite 1.8.0 2022-02-22 [1] CRAN (R 4.2.0)
knitr 1.40 2022-08-24 [1] CRAN (R 4.2.1)
labeling 0.4.2 2020-10-20 [1] CRAN (R 4.2.0)
later 1.3.0 2021-08-18 [1] CRAN (R 4.2.0)
lattice 0.20-45 2021-09-22 [2] CRAN (R 4.2.1)
lifecycle 1.0.2.9000 2022-09-23 [1] Github (r-lib/lifecycle@0a6860a)
lubridate * 1.8.0.9000 2022-05-24 [1] Github (tidyverse/lubridate@0bb49b2)
magick 2.7.3 2021-08-18 [1] CRAN (R 4.2.0)
magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.2.0)
MASS 7.3-58.1 2022-08-03 [2] CRAN (R 4.2.1)
Matrix * 1.5-1 2022-09-13 [1] CRAN (R 4.2.1)
matrixStats 0.62.0 2022-04-19 [1] CRAN (R 4.2.0)
memoise 2.0.1 2021-11-26 [1] CRAN (R 4.2.0)
mime 0.12 2021-09-28 [1] CRAN (R 4.2.0)
miniUI 0.1.1.1 2018-05-18 [1] CRAN (R 4.2.0)
munsell 0.5.0 2018-06-12 [1] CRAN (R 4.2.0)
patchwork * 1.1.2.9000 2022-08-20 [1] Github (thomasp85/patchwork@c14c960)
pillar 1.8.1 2022-08-19 [1] CRAN (R 4.2.1)
pkgbuild 1.3.1 2021-12-20 [1] CRAN (R 4.2.0)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.2.0)
pkgload 1.3.0 2022-06-27 [1] CRAN (R 4.2.1)
png 0.1-7 2013-12-03 [1] CRAN (R 4.2.0)
prettyunits 1.1.1.9000 2022-04-22 [1] Github (r-lib/prettyunits@8706d89)
processx 3.7.0 2022-07-07 [1] CRAN (R 4.2.1)
profvis 0.3.7 2020-11-02 [1] CRAN (R 4.2.0)
promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.2.0)
ps 1.7.1 2022-06-18 [1] CRAN (R 4.2.0)
purrr * 0.9000.0.9000 2022-09-24 [1] Github (tidyverse/purrr@4ab13f5)
R.cache 0.16.0 2022-07-21 [1] CRAN (R 4.2.1)
R.methodsS3 1.8.2 2022-06-13 [1] CRAN (R 4.2.0)
R.oo 1.25.0 2022-06-12 [1] CRAN (R 4.2.0)
R.utils 2.12.0 2022-06-28 [1] CRAN (R 4.2.1)
R6 2.5.1.9000 2022-08-04 [1] Github (r-lib/R6@87d5e45)
ragg 1.2.2.9000 2022-09-12 [1] Github (r-lib/ragg@904e145)
RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.2.0)
Rcpp 1.0.9 2022-07-08 [1] CRAN (R 4.2.1)
readr * 2.1.2.9000 2022-09-20 [1] Github (tidyverse/readr@5cac6ed)
readxl 1.4.1.9000 2022-09-15 [1] Github (tidyverse/readxl@1835c96)
remotes 2.4.2 2022-09-12 [1] Github (r-lib/remotes@bc0949d)
reticulate 1.26 2022-08-31 [1] CRAN (R 4.2.1)
rjson 0.2.21 2022-01-09 [1] CRAN (R 4.2.0)
rlang 1.0.6 2022-09-24 [1] Github (r-lib/rlang@66454bd)
rmarkdown 2.16.1 2022-09-24 [1] Github (rstudio/rmarkdown@9577707)
Rttf2pt1 1.3.10 2022-02-07 [1] CRAN (R 4.2.0)
S4Vectors 0.34.0 2022-04-26 [1] Bioconductor
sass 0.4.2 2022-07-16 [1] CRAN (R 4.2.1)
scales 1.2.1.9000 2022-08-20 [1] Github (r-lib/scales@b3df2fb)
sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.2.0)
shape 1.4.6 2021-05-19 [1] CRAN (R 4.2.0)
shiny 1.7.2 2022-07-19 [1] CRAN (R 4.2.1)
stringi 1.7.8 2022-07-11 [1] CRAN (R 4.2.1)
stringr * 1.4.1.9000 2022-08-21 [1] Github (tidyverse/stringr@792bc92)
styler * 1.7.0.9002 2022-09-21 [1] Github (r-lib/styler@1f4437b)
systemfonts 1.0.4 2022-02-11 [1] CRAN (R 4.2.0)
textshaping 0.3.6 2021-10-13 [1] CRAN (R 4.2.0)
tibble * 3.1.8.9002 2022-09-24 [1] Github (tidyverse/tibble@e9db4f4)
tidyr * 1.2.1.9000 2022-09-09 [1] Github (tidyverse/tidyr@653def2)
tidyselect 1.1.2.9000 2022-09-21 [1] Github (r-lib/tidyselect@edd0a3b)
tidyverse * 1.3.2.9000 2022-09-12 [1] Github (tidyverse/tidyverse@3be8283)
tzdb 0.3.0 2022-03-28 [1] CRAN (R 4.2.0)
urlchecker 1.0.1 2021-11-30 [1] CRAN (R 4.2.0)
usethis 2.1.6.9000 2022-09-23 [1] Github (r-lib/usethis@8ecb7ab)
utf8 1.2.2 2021-07-24 [1] CRAN (R 4.2.0)
vctrs 0.4.1.9000 2022-09-19 [1] Github (r-lib/vctrs@0a219ba)
vroom 1.5.7.9000 2022-09-09 [1] Github (r-lib/vroom@0c2423e)
wesanderson 0.3.6.9000 2022-04-22 [1] Github (karthik/wesanderson@2796b59)
withr 2.5.0 2022-03-03 [1] CRAN (R 4.2.0)
xfun 0.33 2022-09-12 [1] CRAN (R 4.2.1)
xtable 1.8-4 2019-04-21 [1] CRAN (R 4.2.0)
yaml 2.3.5 2022-02-21 [1] CRAN (R 4.2.0)
[1] /opt/homebrew/lib/R/4.2/site-library
[2] /opt/homebrew/Cellar/r/4.2.1_4/lib/R/library
─ Python configuration ───────────────────────────────────────────────────────
python: /Users/jialei/.pyenv/shims/python
libpython: /Users/jialei/.pyenv/versions/mambaforge-4.10.3-10/lib/libpython3.9.dylib
pythonhome: /Users/jialei/.pyenv/versions/mambaforge-4.10.3-10:/Users/jialei/.pyenv/versions/mambaforge-4.10.3-10
version: 3.9.13 | packaged by conda-forge | (main, May 27 2022, 17:00:33) [Clang 13.0.1 ]
numpy: /Users/jialei/.pyenv/versions/mambaforge-4.10.3-10/lib/python3.9/site-packages/numpy
numpy_version: 1.22.4
numpy: /Users/jialei/.pyenv/versions/mambaforge-4.10.3-10/lib/python3.9/site-packages/numpy
NOTE: Python version was forced by RETICULATE_PYTHON
──────────────────────────────────────────────────────────────────────────────Citation
@article{yu,
author = {Leqian Yu and Yulei Wei and Jialei Duan and Daniel A.
Schmitz and Masahiro Sakurai and Lei Wang and Kunhua Wang and Shuhua
Zhao and Gary C. Hon and Jun Wu},
editor = {},
publisher = {Nature Publishing Group},
title = {Blastocyst-Like Structures Generated from Human Pluripotent
Stem Cells},
journal = {Nature},
volume = {591},
number = {7851},
pages = {620 - 626},
date = {},
url = {https://doi.org/10.1038/s41586-021-03356-y},
doi = {10.1038/s41586-021-03356-y},
langid = {en},
abstract = {Human blastoids provide a readily accessible, scalable,
versatile and perturbable alternative to blastocysts for studying
early human development, understanding early pregnancy loss and
gaining insights into early developmental defects.}
}