
Sys.time()[1] "2023-09-10 03:24:39 CDT"Sys.time()[1] "2023-09-10 03:24:39 CDT"[1] "America/Chicago"PROJECT_DIR <- file.path(
"/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation"
)Load required packages.
library(tidyverse)
## ── Attaching core tidyverse packages ─────────────────── tidyverse 2.0.0.9000 ──
## ✔ dplyr 1.1.3 ✔ readr 2.1.4.9000
## ✔ forcats 1.0.0.9000 ✔ stringr 1.5.0.9000
## ✔ ggplot2 3.4.3.9000 ✔ tibble 3.2.1.9005
## ✔ lubridate 1.9.2.9000 ✔ tidyr 1.3.0.9000
## ✔ purrr 1.0.2.9000
## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## ✖ dplyr::filter() masks stats::filter()
## ✖ dplyr::lag() masks stats::lag()
## ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
library(Matrix)
##
## Attaching package: 'Matrix'
##
## The following objects are masked from 'package:tidyr':
##
## expand, pack, unpack
library(patchwork)
library(extrafont)
## Registering fonts with R`%+replace%` <- ggplot2::`%+replace%`add_panel_border <- function() {
ggplot2::theme(
plot.background = element_rect(
colour = "grey70", fill = NA, linewidth = 0.25
)
)
}plot_embedding_highlight <- function(embedding, x, y, label,
geom_point_size = 0.2,
n_cols = 3) {
cell_metadata_selected <- x
selected_column <- y
purrr::map(levels(cell_metadata_selected[[selected_column]]), \(x) {
values <- embedding |>
dplyr::left_join(cell_metadata_selected, by = "cell") |>
dplyr::mutate(
value = case_when(
.data[[selected_column]] == x ~ "1",
.data[[selected_column]] != x ~ "0"
)
) |>
dplyr::pull(value) |>
as.integer() |>
as.factor()
plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{label}; ",
"{x}: {sum(as.integer(as.character(values)), na.rm = TRUE)}"
),
color_labels = FALSE,
color_legend = FALSE,
sort_values = TRUE,
shuffle_values = FALSE,
rasterise = RASTERISED,
geom_point_size = geom_point_size
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
na.translate = TRUE,
values = c("#7F7F7F", "salmon"),
na.value = "grey70"
) +
ggplot2::annotate(
geom = "text",
x = Inf,
y = Inf,
label = sum(as.integer(as.character(values)), na.rm = TRUE),
size = 5 / ggplot2::.pt,
hjust = 1,
vjust = 1,
na.rm = FALSE
)
}) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = n_cols) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
}np <- reticulate::import("numpy", convert = TRUE)
# scipy.sparse <- reticulate::import(module = "scipy.sparse", convert = TRUE)
cat("numpy version:", np$`__version__`, "\n")numpy version: 1.24.3 reticulate::py_config()python: /Users/jialei/.pyenv/shims/python
libpython: /Users/jialei/.pyenv/versions/mambaforge-22.9.0-3/lib/libpython3.10.dylib
pythonhome: /Users/jialei/.pyenv/versions/mambaforge-22.9.0-3:/Users/jialei/.pyenv/versions/mambaforge-22.9.0-3
version: 3.10.9 | packaged by conda-forge | (main, Feb 2 2023, 20:26:08) [Clang 14.0.6 ]
numpy: /Users/jialei/.pyenv/versions/mambaforge-22.9.0-3/lib/python3.10/site-packages/numpy
numpy_version: 1.24.3
numpy: /Users/jialei/.pyenv/versions/mambaforge-22.9.0-3/lib/python3.10/site-packages/numpy
NOTE: Python version was forced by RETICULATE_PYTHONembedding <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"clustering",
"LW60_LW61_LW119_LW120_LW121_LW122_PRJEB11202_PRJNA562548_PRJEB40781_PRJNA555602_PRJNA720968",
"exploring",
CLUSTERING_METHOD,
EMBEDDING_FILE
),
show_col_types = FALSE
) |>
dplyr::mutate(
batch = dplyr::case_when(
batch == "GSM4734573" ~ "PRJNA658478",
TRUE ~ batch
),
bioproject = dplyr::case_when(
batch %in% c("LW60", "LW61") ~ "PRJNA632839",
#
batch %in% c("GSM3956280", "GSM3956281") ~ "PRJNA555602",
batch %in% c("GSM4734573") ~ "PRJNA658478",
#
batch %in% c(
"GSM4816780", "GSM4816781", "GSM4816782"
) ~ "PRJNA667174",
batch %in% c("GSM5387817", "GSM5387818") ~ "PRJNA738498",
TRUE ~ batch
)
)# PRJNA632839 Yu et al. 2021
adata_files <- purrr::map(c("LW36_LW58_LW59_LW60_LW61"), \(x) {
file.path(
PROJECT_DIR,
"raw",
"PRJNA632839",
x,
"matrix",
"adata.h5ad"
)
})
# PRJNA658478 Liu et al. 2021
# PRJNA720968 Yanagida et al. 2021
# PRJNA737139 Kagawa et al. 2021
##
# PRJNA667174 Fan et al. 2021
# PRJNA738498 Sozen et al. 2021
adata_files <- c(
adata_files,
purrr::map(c(
"PRJNA658478",
"PRJNA720968",
"PRJNA737139",
"PRJNA667174",
"PRJNA738498"
), \(x) {
file.path(
PROJECT_DIR,
"raw",
"public",
x,
"matrix",
"adata.h5ad"
)
})
)
purrr::map_lgl(adata_files, file.exists)[1] TRUE TRUE TRUE TRUE TRUE TRUEadata_files <- c(
adata_files,
purrr::map(c("LW119", "LW120", "LW121", "LW122"), \(x) {
file.path(
PROJECT_DIR,
"raw",
x,
"matrix",
"adata.h5ad"
)
})
)
# Petropoulos et al. 2016; PRJEB11202
# Zhou et al. 2019; PRJNA431392
# Xiang et al. 2020; PRJNA562548
# Tyser et al. 2021; PRJEB40781
# Zheng et al. 2019; PRJNA555602
adata_files <- c(
adata_files,
purrr::map(
c(
"PRJEB11202",
"PRJNA431392",
"PRJNA562548",
"PRJEB40781",
"PRJNA555602"
), \(x) {
file.path(
PROJECT_DIR,
"raw",
"public",
x,
"matrix",
"adata.h5ad"
)
}
)
)
purrr::map_lgl(adata_files, file.exists) [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUEBACKED <- NULL
matrix_readcount_use <- purrr::map(adata_files, function(x) {
cat(x, "\n")
ad$read_h5ad(
filename = x, backed = BACKED
) |>
# convert_adata(cells_selected = embedding$cell)
extract_matrix_from_adata(cells_selected = embedding$cell)
}) |>
purrr::reduce(cbind)/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/PRJNA632839/LW36_LW58_LW59_LW60_LW61/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJNA658478/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJNA720968/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJNA737139/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJNA667174/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJNA738498/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/LW119/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/LW120/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/LW121/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/LW122/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJEB11202/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJNA431392/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJNA562548/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJEB40781/matrix/adata.h5ad
/Users/jialei/Dropbox/Data/Projects/UTSW/Peri-implantation/raw/public/PRJNA555602/matrix/adata.h5ad if (FALSE) {
adata_files[[1]]
adata <- ad$read_h5ad(
filename = adata_files[[1]], backed = BACKED
)
m <- Matrix::sparseMatrix(
i = as.integer(adata$X$indices),
p = as.integer(adata$X$indptr),
x = as.numeric(adata$X$data),
dims = unlist(adata$shape),
dimnames = list(
rownames(adata$obs),
rownames(adata$var)
),
index1 = FALSE
)
}
matrix_readcount_use <- matrix_readcount_use[
,
sort(colnames(matrix_readcount_use))
]
matrix_readcount_use |> dim()[1] 33538 31481BACKED <- "r"
cell_metadata <- purrr::map(adata_files, function(x) {
ad$read_h5ad(
filename = x, backed = BACKED
)$obs |>
tibble::rownames_to_column(var = "cell") |>
dplyr::select(cell, everything())
# |> dplyr::select(cell : mt_percentage)
}) |>
dplyr::bind_rows()embedding |>
dplyr::left_join(
cell_metadata |>
dplyr::select(cell, num_umis),
by = "cell"
) |>
dplyr::group_by(batch) |>
dplyr::summarise(
num_cells = n(),
meidan_umis = median(num_umis)
) |>
gt::gt()| batch | num_cells | meidan_umis |
|---|---|---|
| GSM3956280 | 5454 | 23322.0 |
| GSM3956281 | 4512 | 27887.0 |
| LW119 | 2236 | 2956.5 |
| LW120 | 435 | 17392.0 |
| LW121 | 5130 | 12536.0 |
| LW122 | 287 | 100092.0 |
| LW60 | 4497 | 14421.0 |
| LW61 | 5156 | 7625.0 |
| PRJEB11202 | 1529 | 1551093.0 |
| PRJEB40781 | 1195 | 899241.0 |
| PRJNA562548 | 555 | 10165004.0 |
| PRJNA720968 | 495 | 7690661.0 |
Check memory usage.
walk(list(matrix_readcount_use, embedding), \(x) {
print(object.size(x), units = "auto", standard = "SI")
})1.5 GB
6.7 MBstudies <- tibble::tribble(
~bioproject, ~citation,
"PRJNA277181", "Blakeley et al. 2015",
"PRJEB11202", "Petropoulos et al. 2016",
"PRJNA431392", "Zhou et al. 2019",
"PRJNA562548", "Xiang et al. 2020",
#
"PRJEB40781", "Tyser et al. 2021",
#
"PRJNA555602", "Zheng et al. 2019",
#
"PRJNA632839", "Yu et al. 2021",
"PRJNA658478", "Liu et al. 2021",
"PRJNA720968", "Yanagida et al. 2021",
"PRJNA667174", "Fan et al. 2021",
"PRJNA738498", "Sozen et al. 2021",
"PRJNA737139", "Kagawa et al. 2021"
)
studies <- setNames(
object = studies$citation,
nm = studies$bioproject
)
embedding <- embedding |>
dplyr::mutate(
study = studies[bioproject],
study = dplyr::case_when(
is.na(study) ~ "This study",
TRUE ~ as.character(study)
),
study = factor(
study,
levels = c(
"This study",
"Yu et al. 2021",
"Yanagida et al. 2021",
"Petropoulos et al. 2016",
"Xiang et al. 2020",
"Tyser et al. 2021",
"Zheng et al. 2019"
)
),
batch_annotated = paste(study, batch, sep = "; ")
)embedding |>
dplyr::left_join(
cell_metadata |> dplyr::select(cell, num_umis),
by = "cell"
) |>
dplyr::group_by(study, batch) |>
dplyr::summarise(
num_cells = n(),
median_umis = median(num_umis)
) |>
gt::gt() |>
gt::data_color(
columns = c(median_umis),
fn = scales::col_numeric(
palette = c(
"green", "orange", "red"
),
domain = NULL
)
) |>
gt::fmt_number(
columns = c(median_umis),
sep_mark = ",",
decimals = 1,
use_seps = TRUE,
suffixing = FALSE
) |>
gt::fmt_number(
columns = c(num_cells),
sep_mark = ",",
decimals = 0,
use_seps = TRUE,
suffixing = FALSE
) |>
gt::grand_summary_rows(
columns = c(num_cells),
fns = list(
Sum = ~ sum(.)
),
fmt = ~ gt::fmt_number(., decimals = 0, use_seps = TRUE)
)`summarise()` has grouped output by 'study'. You can override using the
`.groups` argument.| batch | num_cells | median_umis | |
|---|---|---|---|
| This study | |||
| LW119 | 2,236 | 2,956.5 | |
| LW120 | 435 | 17,392.0 | |
| LW121 | 5,130 | 12,536.0 | |
| LW122 | 287 | 100,092.0 | |
| Yu et al. 2021 | |||
| LW60 | 4,497 | 14,421.0 | |
| LW61 | 5,156 | 7,625.0 | |
| Yanagida et al. 2021 | |||
| PRJNA720968 | 495 | 7,690,661.0 | |
| Petropoulos et al. 2016 | |||
| PRJEB11202 | 1,529 | 1,551,093.0 | |
| Xiang et al. 2020 | |||
| PRJNA562548 | 555 | 10,165,004.0 | |
| Tyser et al. 2021 | |||
| PRJEB40781 | 1,195 | 899,241.0 | |
| Zheng et al. 2019 | |||
| GSM3956280 | 5,454 | 23,322.0 | |
| GSM3956281 | 4,512 | 27,887.0 | |
| Sum | — | 31,481 | — |
GEOM_POINT_SIZE <- 0.25
RASTERISED <- TRUE
x_column <- "x_umap_min_dist=0.1"
y_column <- "y_umap_min_dist=0.1"
EMBEDDING_TITLE_PREFIX <- "UMAP"
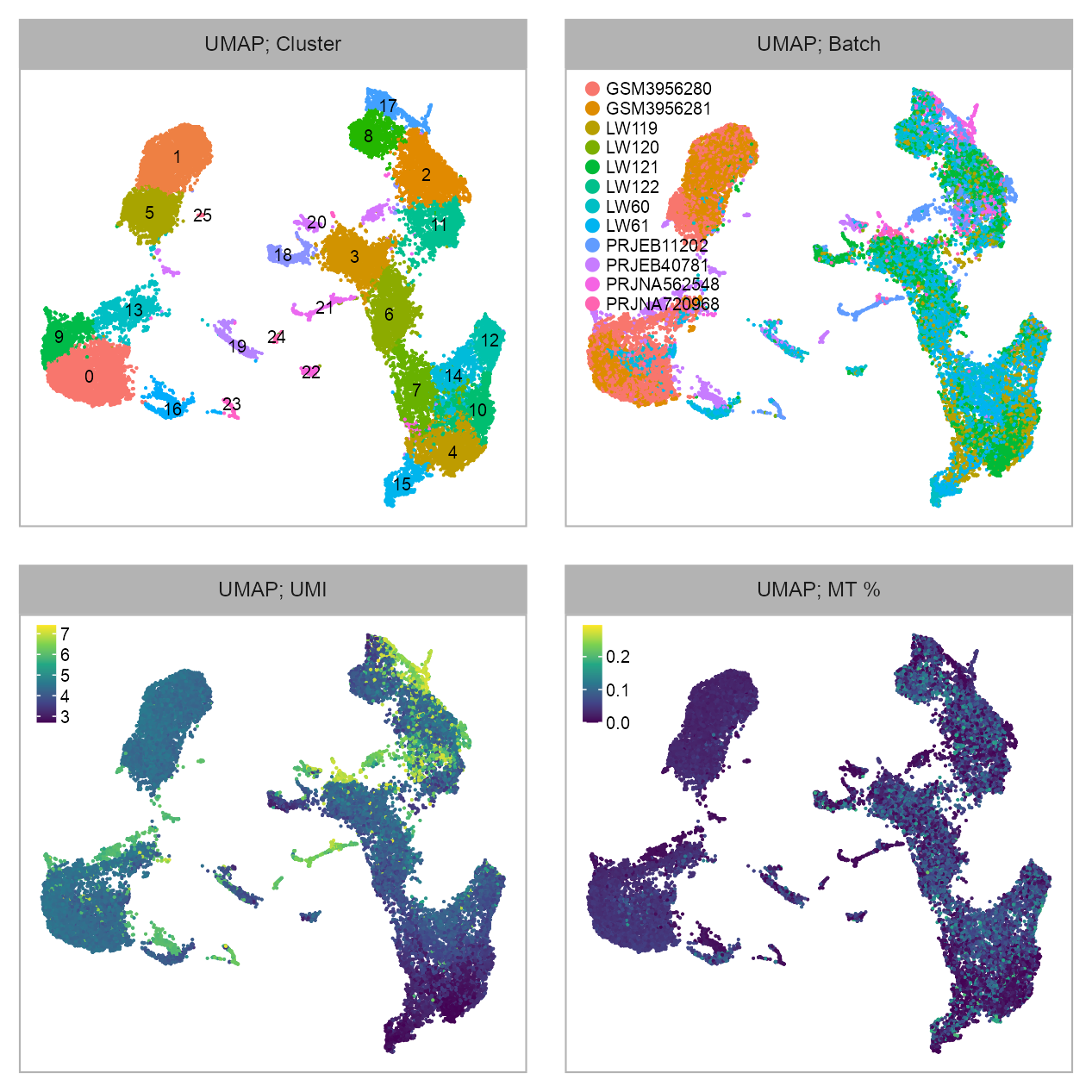
embedding_type <- EMBEDDING_TITLE_PREFIXp_embedding_leiden <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = embedding$leiden |> as.factor(),
label = glue::glue("{EMBEDDING_TITLE_PREFIX}; Cluster"),
color_labels = TRUE,
color_legend = FALSE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2
) +
theme_customized_embedding()
p_embedding_batch <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = embedding$batch |> as.factor(),
label = glue::glue("{EMBEDDING_TITLE_PREFIX}; Batch"),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2
) +
theme_customized_embedding()
p_embedding_UMI <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = log10(Matrix::colSums(matrix_readcount_use[, embedding$cell])),
label = glue::glue("{EMBEDDING_TITLE_PREFIX}; UMI"),
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2
) +
theme_customized_embedding()
p_embedding_MT <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = embedding |>
dplyr::left_join(cell_metadata, by = c("cell")) |>
dplyr::pull(mt_percentage),
label = glue::glue("{EMBEDDING_TITLE_PREFIX}; MT %"),
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2
) +
theme_customized_embedding()
purrr::reduce(
list(
p_embedding_leiden,
p_embedding_batch,
p_embedding_UMI,
p_embedding_MT
), `+`
) +
patchwork::plot_layout(ncol = 2) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
cell_metadata_PRJNA632839 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw",
"PRJNA632839",
"embedding.csv.gz"
),
show_col_types = FALSE
) |>
dplyr::filter(batch %in% c("LW60", "LW61")) |>
dplyr::mutate(
lineage = dplyr::case_when(
louvain %in% c(11) ~ "ELCs",
louvain %in% c(18) ~ "HLCs",
louvain %in% c(0, 1, 8, 9, 12, 17) ~ "TLCs",
TRUE ~ paste0("U", louvain)
),
lineage = factor(
lineage,
levels = stringr::str_sort(unique(lineage), numeric = TRUE)
)
)cell_metadata_PRJNA720968 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw/public",
"PRJNA720968",
"matrix",
"cell_metadata.csv"
),
show_col_types = FALSE
) |>
dplyr::mutate(
origin = factor(
origin,
levels = c("Blastocyst", "Blastoid")
),
#
developmental_stage = factor(
developmental_stage,
levels = stringr::str_sort(
x = unique(developmental_stage), numeric = TRUE
)
),
#
lineage = paste(origin, lineage, sep = ": "),
lineage = factor(
lineage,
levels = c(
"Blastocyst: Epiblast",
"Blastocyst: Hypoblast",
"Blastocyst: Inner Cell Mass",
"Blastocyst: Inner Cell Mass-Trophectoderm Transition",
"Blastocyst: Early Trophectoderm",
"Blastocyst: Trophectoderm",
#
"Blastoid: Epiblast",
"Blastoid: Hypoblast",
"Blastoid: Transitioning",
"Blastoid: Trophectoderm"
)
)
)cell_metadata_PRJEB11202 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw",
"public",
"PRJEB11202",
"matrix/cell_metadata.csv"
),
show_col_types = FALSE
) |>
dplyr::mutate(
developmental_stage = stringr::str_remove(
string = individual,
pattern = "\\..+$"
),
developmental_stage = factor(
developmental_stage,
levels = stringr::str_sort(
unique(developmental_stage),
numeric = TRUE
)
),
#
lineage = factor(
inferred_lineage,
levels = c(
"epiblast",
"primitive_endoderm",
"trophectoderm",
"not_applicable"
)
)
) |>
dplyr::select(
cell, lineage, developmental_stage
)embryos_selected_PRJNA431392 <- c(
"ha_D6_E2",
"hm_D6_E1",
"hm_D6_E2",
"hm_D8_E2",
"hm_D8_E3",
"hm_D8_E5",
"ha_D8_E1",
"hm_D8_E1",
"hv_D8_E1",
"hv_D8_E2",
"hv_D8_E3",
"hv_D10_E6",
"ha_D10_E1",
"ha_D10_E2",
"hm_D10_E4",
"hm_D10_E9",
"hv_D10_E7",
"hv_D10_E8",
"ha_D12_E1",
"hv_D12_E1",
"hv_D12_E2"
)
cell_metadata_PRJNA431392 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw",
"public",
"PRJNA431392",
"matrix/cell_metadata.csv"
),
show_col_types = FALSE
) |>
dplyr::mutate(
developmental_stage = factor(
Day,
levels = stringr::str_sort(
unique(Day),
numeric = TRUE
)
),
lineage = factor(
Lineage,
levels = c(
"EPI",
"PE",
"TE",
"MIX"
)
),
`3184` = case_when(
Ori_Day_Emb %in% embryos_selected_PRJNA431392 ~ "1",
TRUE ~ "0"
)
) |>
dplyr::rename(cell = Sample)
rm(embryos_selected_PRJNA431392)cell_metadata_PRJNA562548 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw",
"public",
"PRJNA562548",
"matrix/cell_metadata.csv"
),
show_col_types = FALSE
) |>
dplyr::select(
cell = `Sample Name`,
developmental_stage = Age,
lineage = Cell_type
) |>
dplyr::mutate(
developmental_stage = stringr::str_replace(
string = developmental_stage,
pattern = "embryo invitro day ",
replacement = "E"
),
developmental_stage = factor(
developmental_stage,
levels = stringr::str_sort(
x = unique(developmental_stage),
numeric = TRUE
)
),
lineage = factor(
lineage,
levels = c(
"ICM",
"EPI",
"PSA-EPI",
"Hypoblast",
"CTBs",
"STBs",
"EVTs"
)
)
)cell_metadata_PRJNA555602 <- vroom::vroom(
file = file.path(
PROJECT_DIR,
"raw",
"public",
"PRJNA555602",
"matrix/cell_metadata.csv"
),
show_col_types = FALSE
) |>
dplyr::mutate(
batch = case_when(
old_ident == "10X_Embryoid" ~ "GSM3956280",
old_ident == "10X_H9_Amnion" ~ "GSM3956281"
),
cell = stringr::str_remove(string = cell, pattern = "\\..+"),
# cell = paste(batch, cell, sep = "_"),
#
cell_type = case_when(
rna_snn_res_0.3 %in% 0 ~ "Transwell-AMLC",
rna_snn_res_0.3 %in% 1 ~ "MeLC2",
rna_snn_res_0.3 %in% 2 ~ "Human_ES_cell",
rna_snn_res_0.3 %in% 3 ~ "MeLC1",
rna_snn_res_0.3 %in% 4 ~ "hPGCLC",
rna_snn_res_0.3 %in% 5 ~ "AMLC"
),
cell_type = factor(
cell_type,
levels = c(
"Human_ES_cell",
# human PGC-like cells
"hPGCLC",
"Transwell-AMLC",
# amniotic ectoderm-like cells
"AMLC",
# mesoderm-like cell
"MeLC1",
"MeLC2"
)
)
)bioproject <- "PRJNA632839"
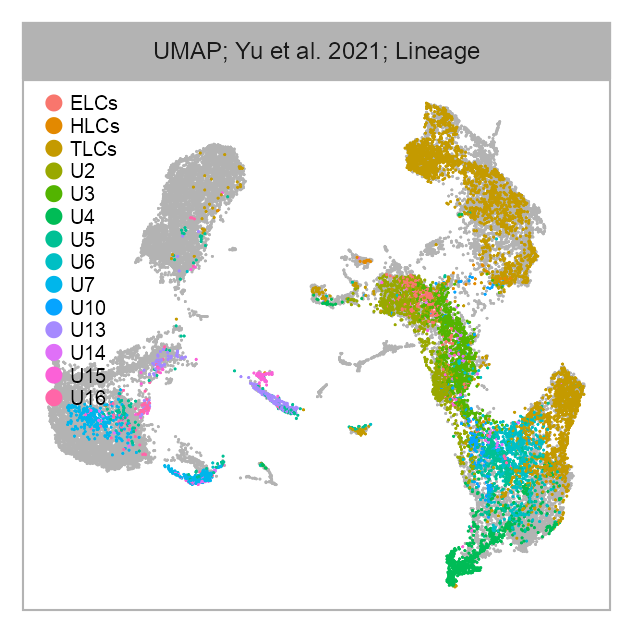
cell_metadata_selected <- cell_metadata_PRJNA632839selected_column <- "lineage"
values <- embedding |>
dplyr::left_join(
cell_metadata_selected |>
dplyr::select(cell, dplyr::all_of(selected_column)),
by = "cell"
) |>
dplyr::pull(.data[[selected_column]])
p_embedding_lineage <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 1.5
) +
theme_customized_embedding()
p_embedding_lineage +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)
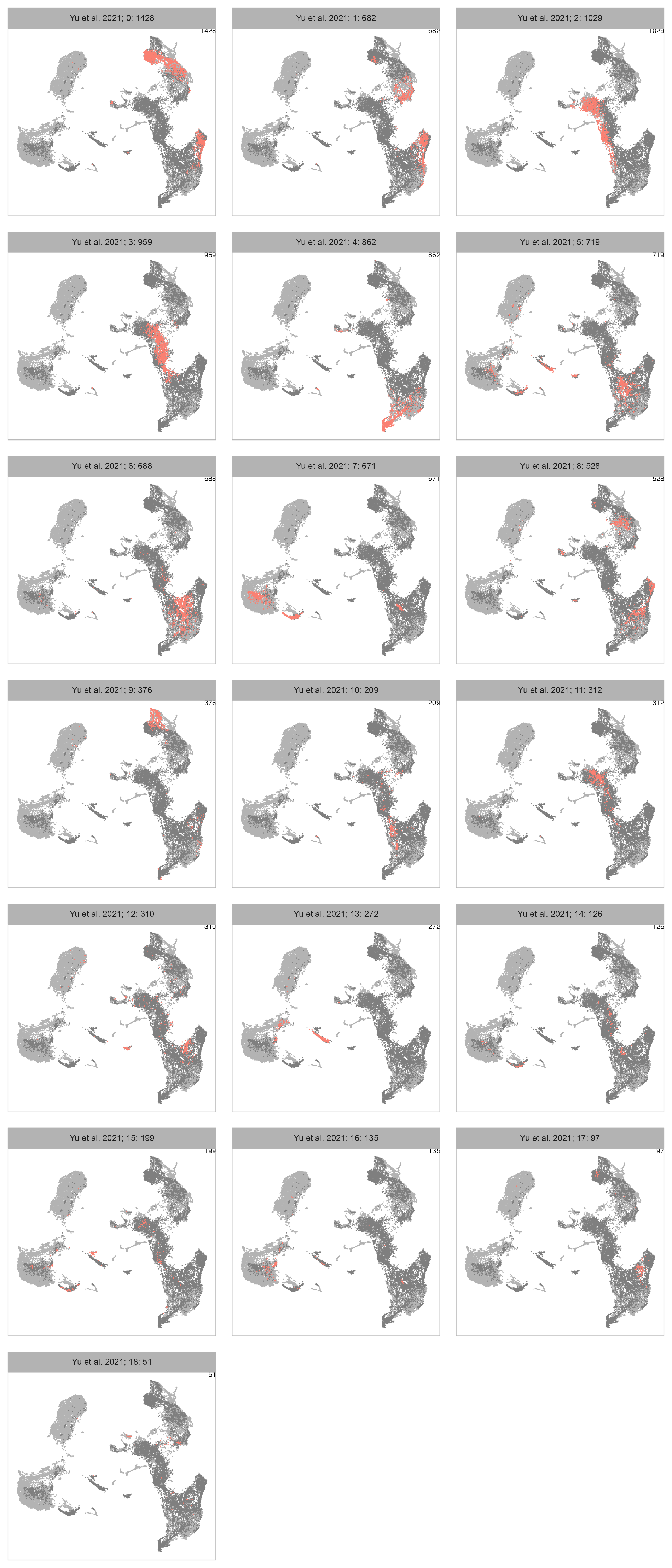
num_columns <- 3
selected_column <- "louvain"
plot_embedding_highlight(
embedding = embedding |>
dplyr::select(!.data[[selected_column]]),
x = cell_metadata_selected |>
dplyr::select(
cell, .data[[selected_column]]
) |>
dplyr::mutate(
louvain = factor(louvain)
),
y = selected_column,
label = studies[bioproject],
geom_point_size = GEOM_POINT_SIZE * 1.5,
n_cols = num_columns
)
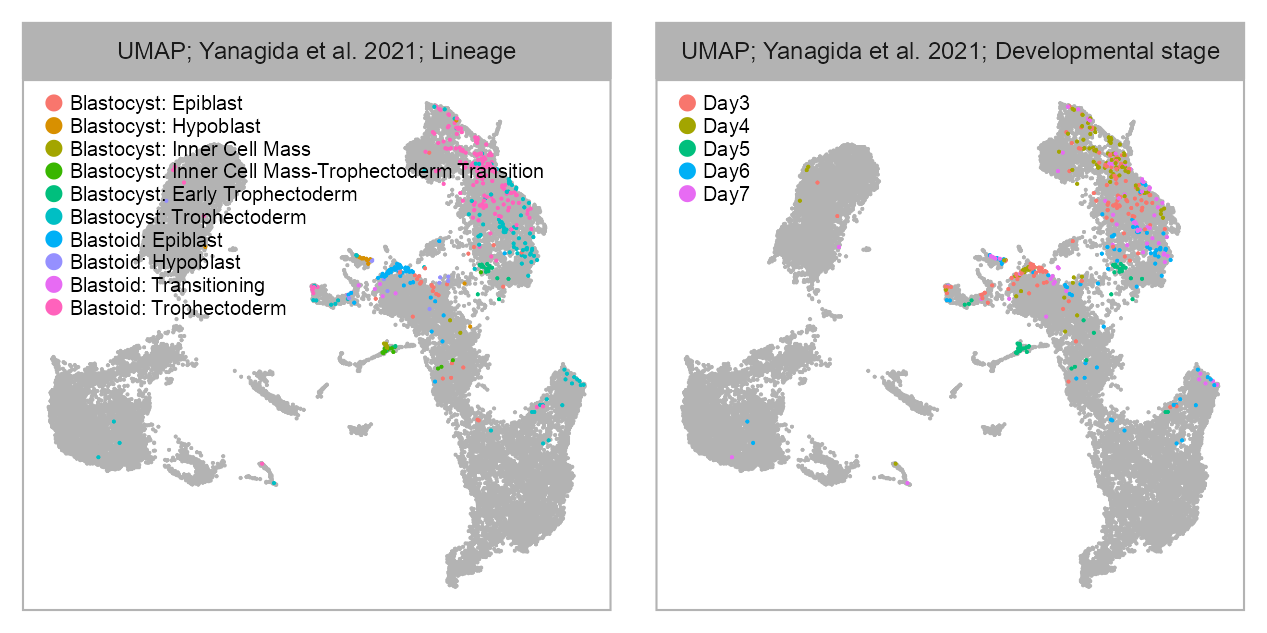
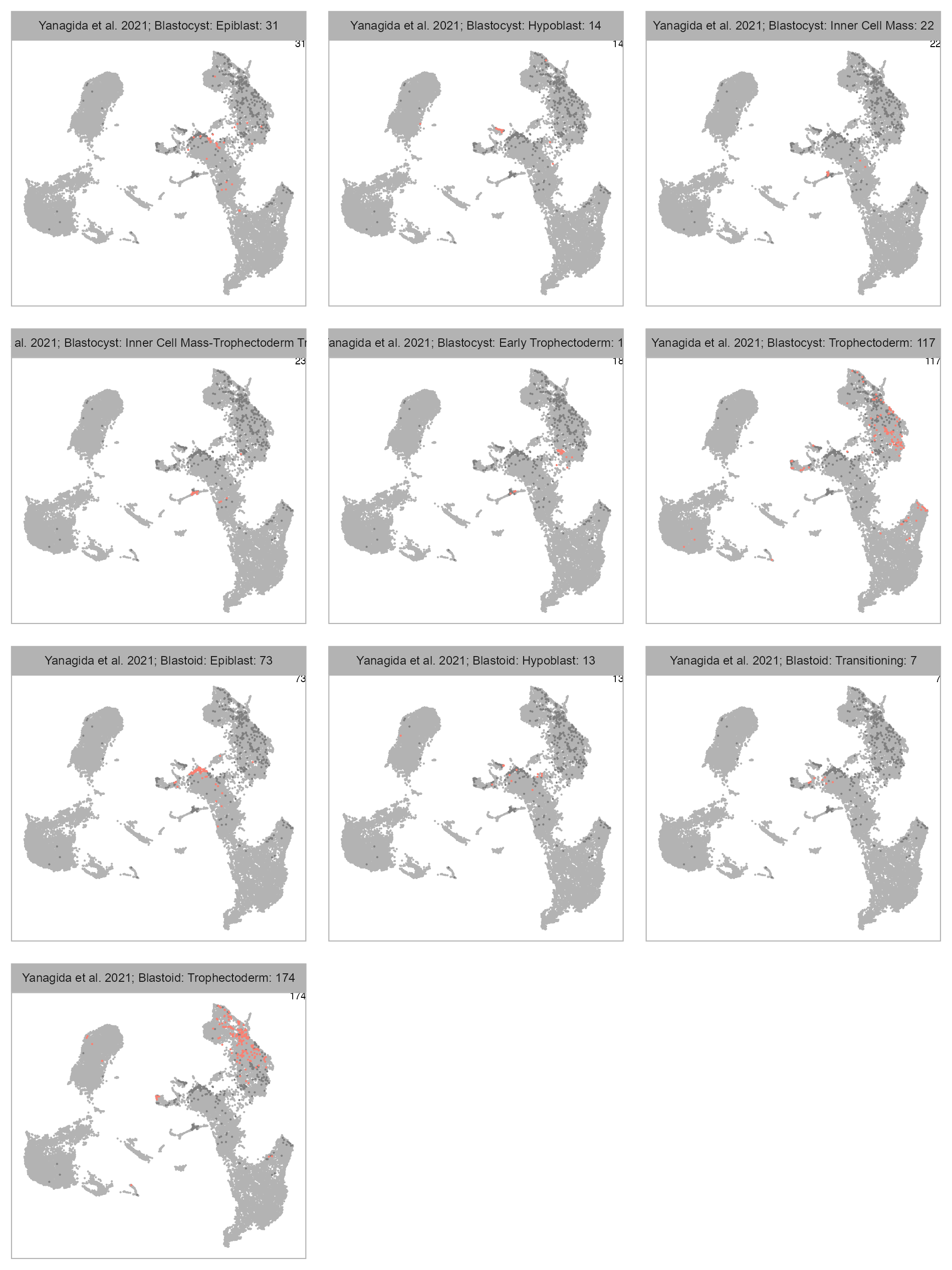
bioproject <- "PRJNA720968"
cell_metadata_selected <- cell_metadata_PRJNA720968selected_column <- "lineage"
values <- embedding |>
dplyr::left_join(
cell_metadata_selected |>
dplyr::select(cell, .data[[selected_column]]),
by = "cell"
) |>
dplyr::pull(.data[[selected_column]])
p_embedding_lineage <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)selected_column <- "developmental_stage"
values <- embedding |>
dplyr::left_join(
cell_metadata_selected |>
dplyr::select(cell, .data[[selected_column]]),
by = "cell"
) |>
dplyr::pull(.data[[selected_column]])
p_embedding_developmental_stage <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)list(
p_embedding_lineage, p_embedding_developmental_stage
) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = 2) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
num_columns <- 3
selected_column <- "lineage"
plot_embedding_highlight(
embedding = embedding,
x = cell_metadata_selected |>
dplyr::select(cell, .data[[selected_column]]),
y = selected_column,
label = studies[bioproject],
geom_point_size = GEOM_POINT_SIZE * 2,
n_cols = num_columns
)
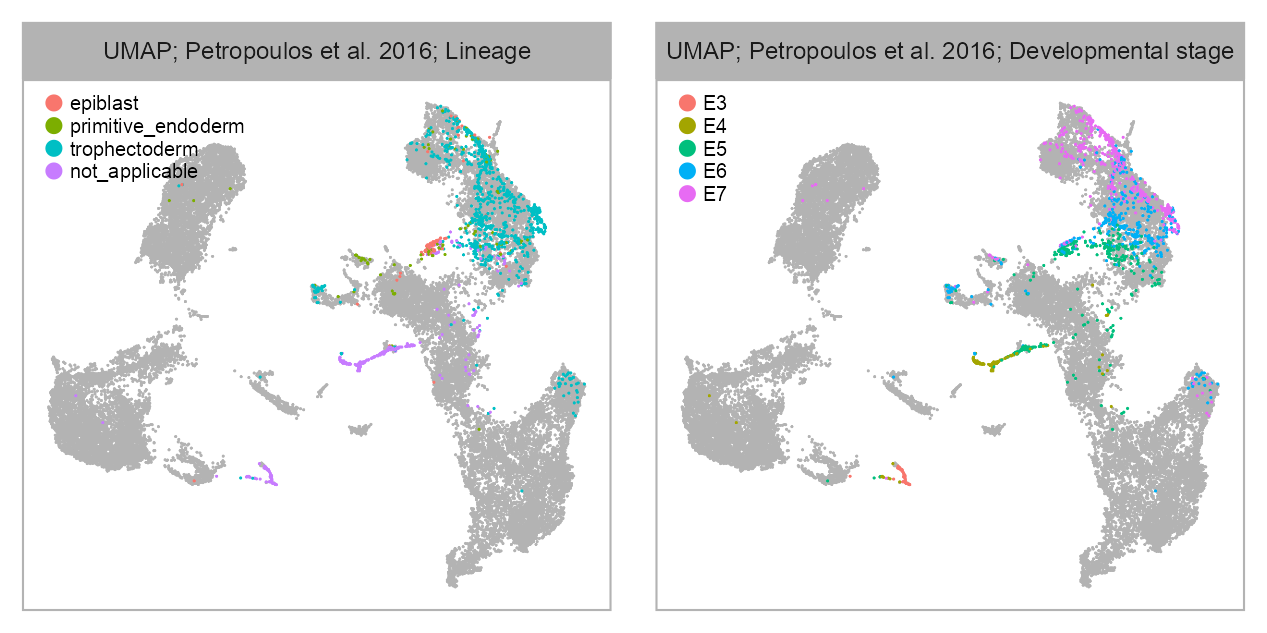
bioproject <- "PRJEB11202"
cell_metadata_selected <- cell_metadata_PRJEB11202selected_column <- "lineage"
values <- embedding |>
dplyr::left_join(cell_metadata_selected, by = "cell") |>
dplyr::pull(.data[[selected_column]])
p_embedding_lineage <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 1.5,
na_value = "grey70"
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)selected_column <- "developmental_stage"
values <- embedding |>
dplyr::left_join(cell_metadata_selected, by = "cell") |>
dplyr::pull(.data[[selected_column]])
p_embedding_developmental_stage <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 1.5,
na_value = "grey70"
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)list(
p_embedding_lineage,
p_embedding_developmental_stage
) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = 2) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
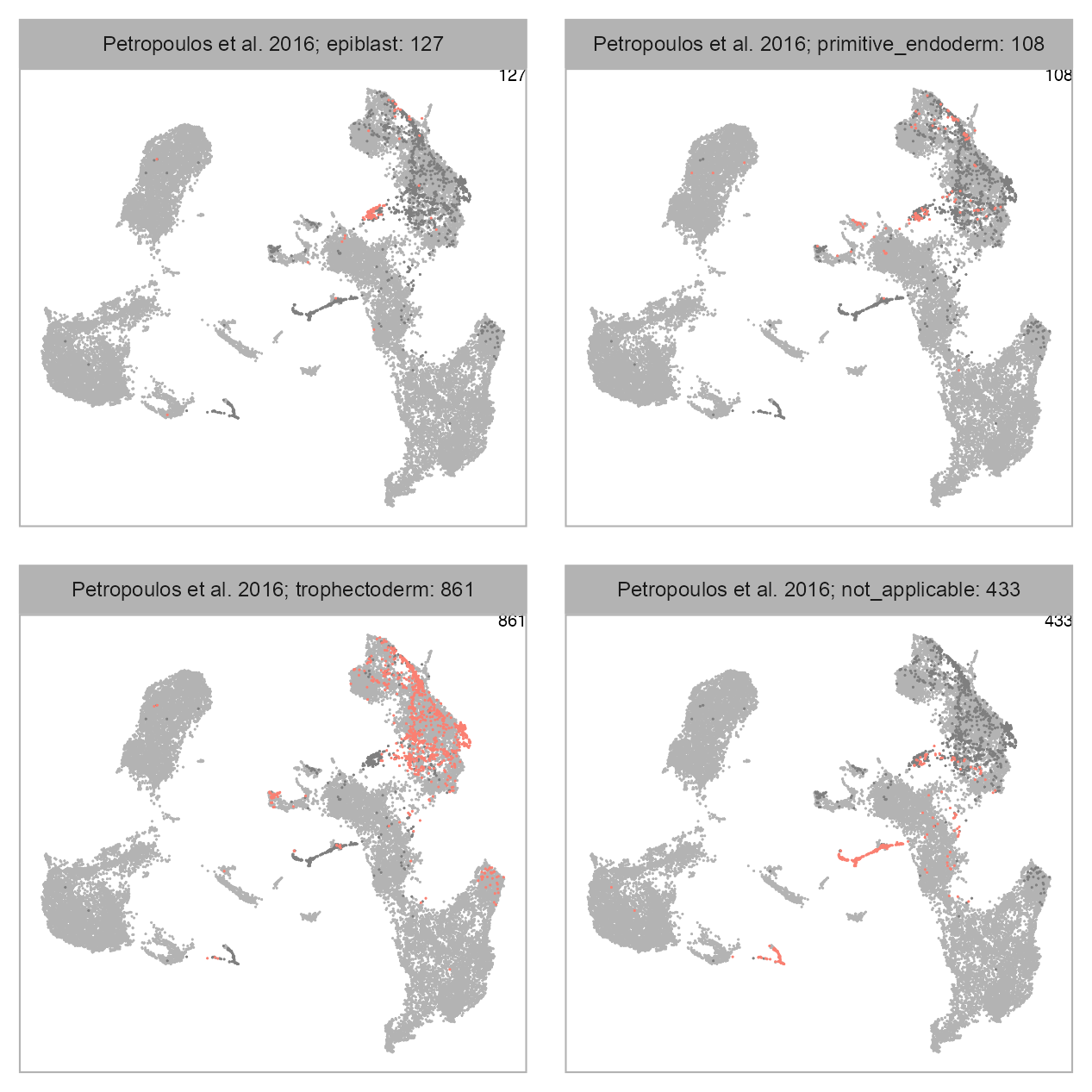
num_columns <- 2
selected_column <- "lineage"
plot_embedding_highlight(
embedding = embedding,
x = cell_metadata_selected,
y = selected_column,
label = studies[bioproject],
geom_point_size = GEOM_POINT_SIZE * 1.5,
n_cols = num_columns
)
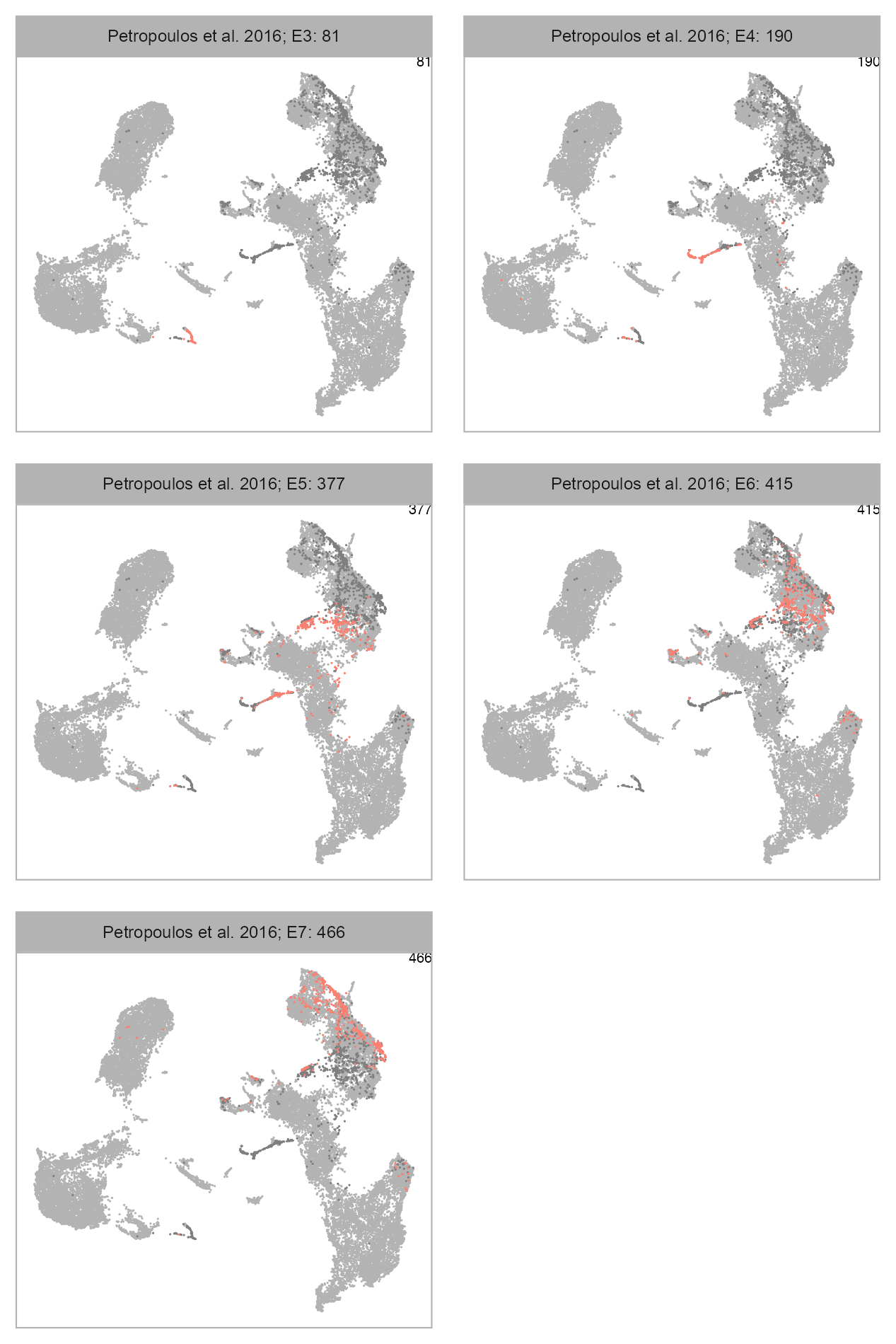
num_columns <- 2
selected_column <- "developmental_stage"
plot_embedding_highlight(
embedding = embedding,
x = cell_metadata_selected,
y = selected_column,
label = studies[bioproject],
geom_point_size = GEOM_POINT_SIZE * 1.5,
n_cols = num_columns
)
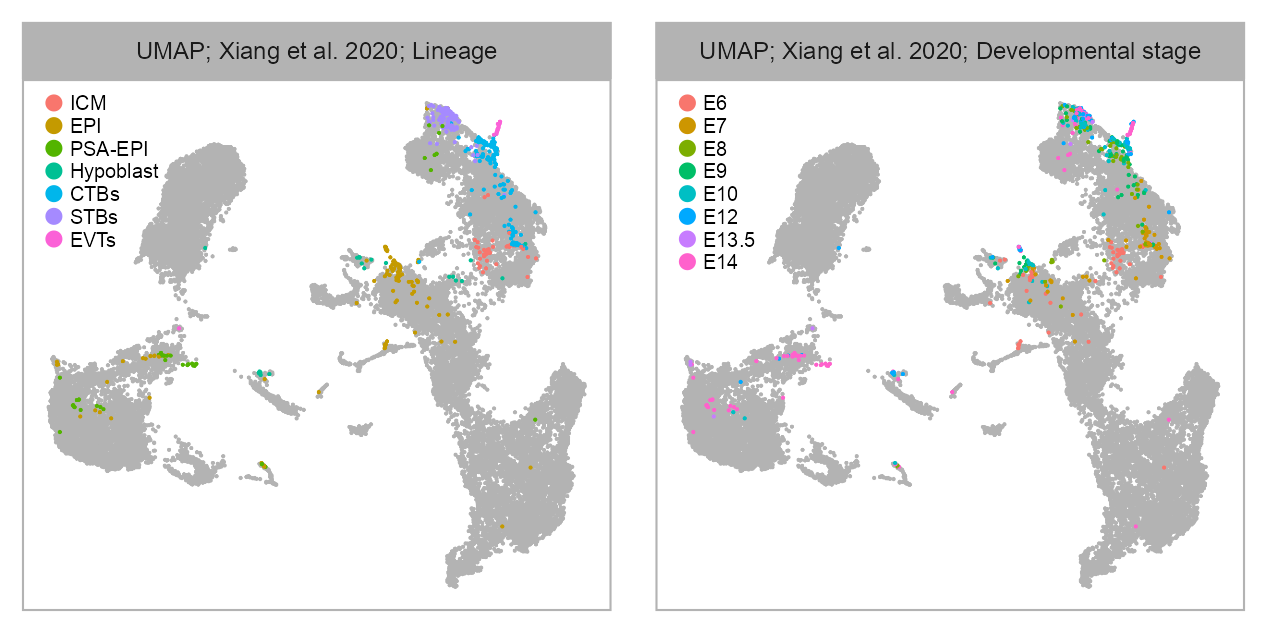
bioproject <- "PRJNA562548"
cell_metadata_selected <- cell_metadata_PRJNA562548selected_column <- "lineage"
values <- embedding |>
dplyr::left_join(cell_metadata_selected, by = "cell") |>
dplyr::pull(.data[[selected_column]])
p_embedding_lineage <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2,
na_value = "grey70"
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)selected_column <- "developmental_stage"
values <- embedding |>
dplyr::left_join(cell_metadata_selected, by = "cell") |>
dplyr::pull(.data[[selected_column]])
p_embedding_developmental_stage <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2,
na_value = "grey70"
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)list(
p_embedding_lineage,
p_embedding_developmental_stage
) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = 2) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
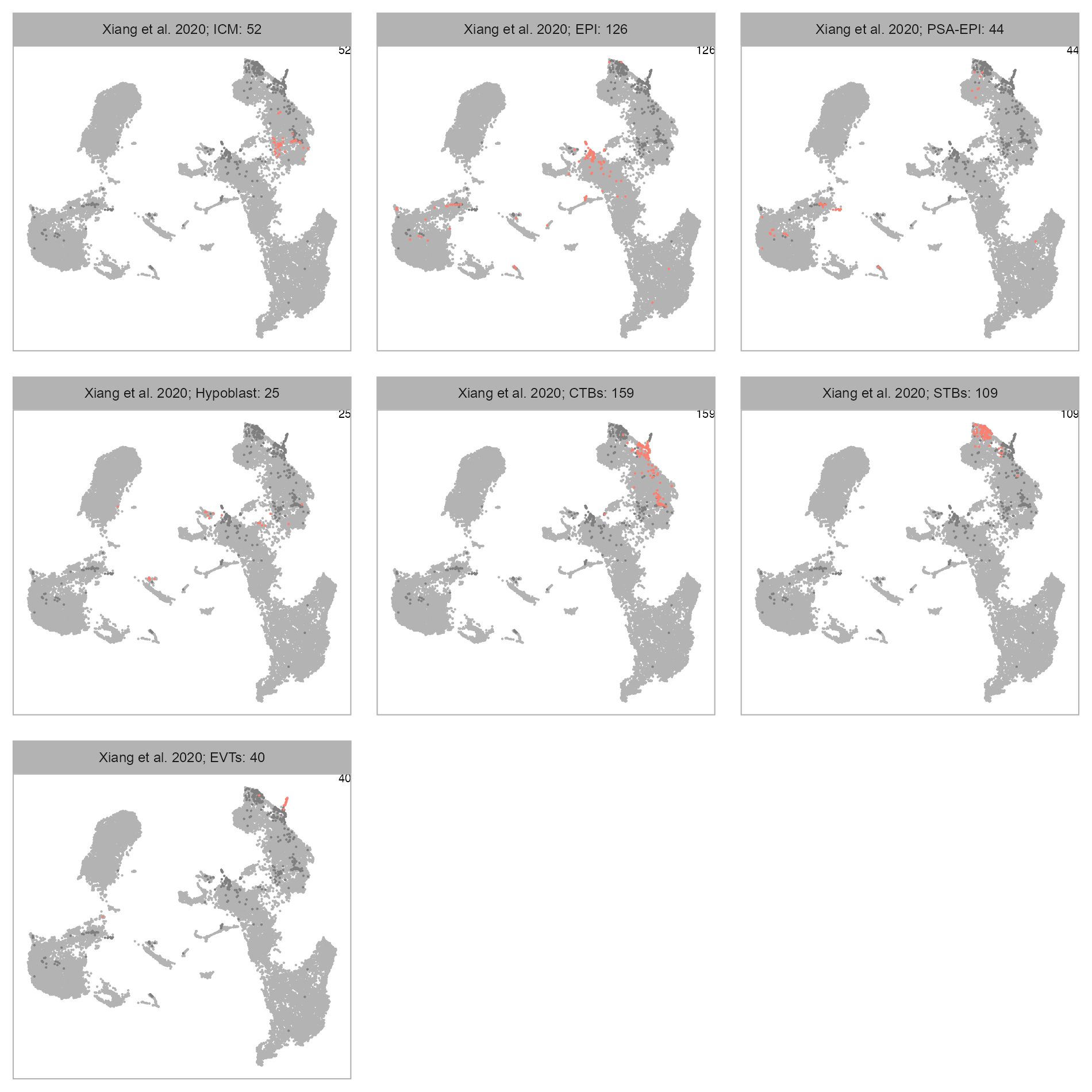
num_columns <- 3
selected_column <- "lineage"
plot_embedding_highlight(
embedding = embedding,
x = cell_metadata_selected,
y = selected_column,
label = studies[bioproject],
geom_point_size = GEOM_POINT_SIZE * 2,
n_cols = num_columns
)
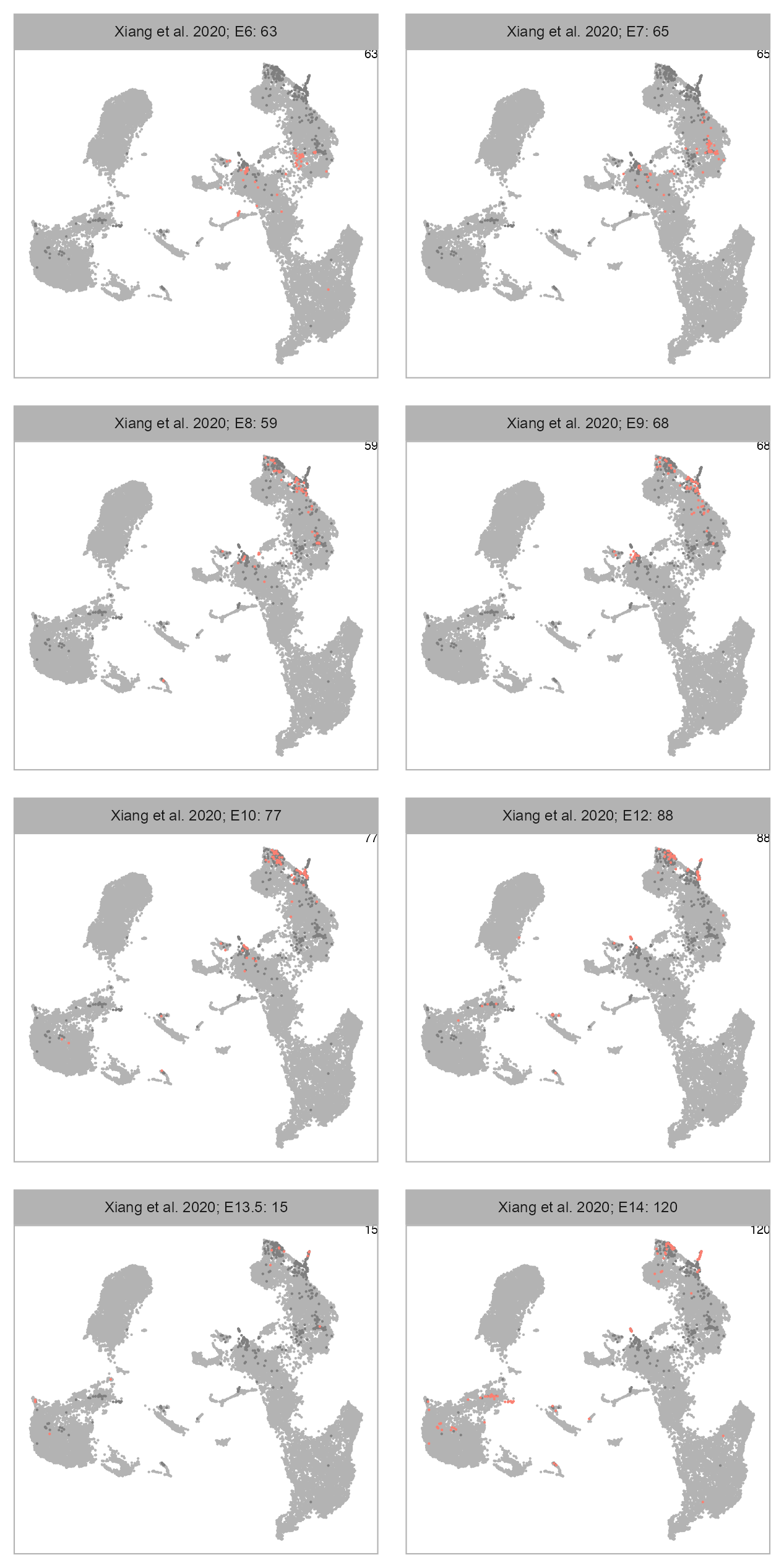
num_columns <- 2
selected_column <- "developmental_stage"
plot_embedding_highlight(
embedding = embedding,
x = cell_metadata_selected,
y = selected_column,
label = studies[bioproject],
geom_point_size = GEOM_POINT_SIZE * 2,
n_cols = num_columns
)
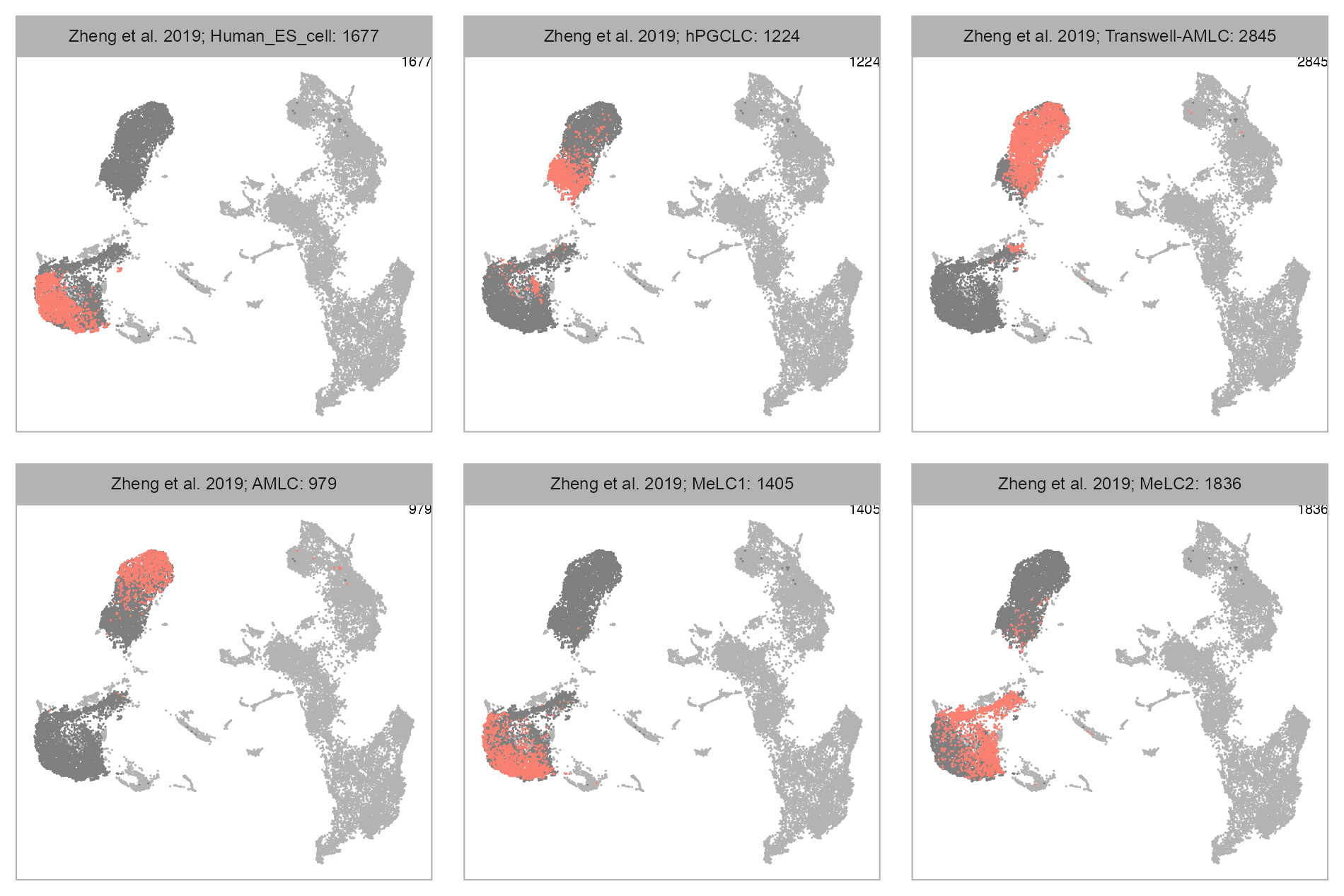
bioproject <- "PRJNA555602"
cell_metadata_selected <- cell_metadata_PRJNA555602selected_column <- "cell_type"
values <- embedding |>
dplyr::left_join(cell_metadata_selected, by = c("cell")) |>
dplyr::pull(.data[[selected_column]])
p_embedding_cell_type <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)num_columns <- 3
selected_column <- "cell_type"
plot_embedding_highlight(
embedding = embedding,
x = cell_metadata_selected,
y = selected_column,
label = studies[bioproject],
geom_point_size = GEOM_POINT_SIZE * 1.5,
n_cols = num_columns
)
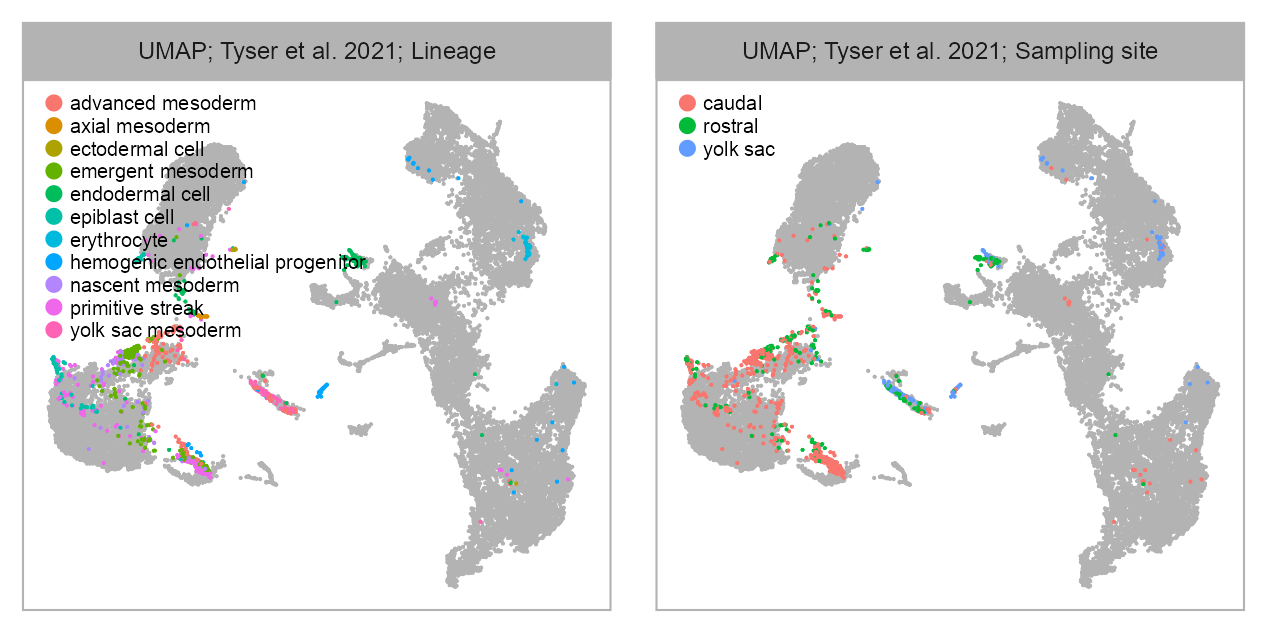
bioproject <- "PRJEB40781"
cell_metadata_selected <- cell_metadata_PRJEB40781selected_column <- "lineage"
values <- embedding |>
dplyr::left_join(cell_metadata_selected, by = "cell") |>
dplyr::pull(.data[[selected_column]])
p_embedding_lineage <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = TRUE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2,
na_value = "grey70"
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)selected_column <- "sampling_site"
values <- embedding |>
dplyr::left_join(cell_metadata_selected, by = "cell") |>
dplyr::pull(.data[[selected_column]])
p_embedding_sampling_site <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; {studies[bioproject]}; ",
selected_column |>
stringr::str_to_title() |>
stringr::str_replace(pattern = "_", replacement = " ")
),
color_labels = FALSE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2,
na_value = "grey70"
) +
theme_customized_embedding() +
ggplot2::scale_color_manual(
values = scales::hue_pal()(n = nlevels(values)),
na.value = "grey70",
breaks = levels(values)
)list(
p_embedding_lineage,
p_embedding_sampling_site
) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = 2) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
embedding <- embedding |>
dplyr::left_join(
cell_metadata_PRJEB11202 |> dplyr::select(cell, lineage),
by = c("cell")
) |>
dplyr::mutate(
lineage = dplyr::case_when(
lineage == "epiblast" ~ "EPI",
lineage == "primitive_endoderm" ~ "HYP",
lineage == "trophectoderm" ~ "TE",
lineage == "not_applicable" ~ "Pre-lineage"
)
) |>
dplyr::left_join(
cell_metadata_PRJNA562548 |>
dplyr::filter(
developmental_stage %in% c("E6", "E7")
) |>
dplyr::select(cell, lineage_ = lineage),
by = "cell"
) |>
dplyr::mutate(
lineage = dplyr::case_when(
lineage_ == "EPI" ~ "EPI",
lineage_ == "Hypoblast" ~ "HYP",
lineage_ == "CTBs" ~ "TE",
lineage_ == "ICM" ~ "ICM",
TRUE ~ lineage
)
) |>
dplyr::select(!lineage_) |>
dplyr::left_join(
cell_metadata_PRJNA720968 |>
dplyr::filter(origin == "Blastocyst") |>
dplyr::select(cell, lineage_ = lineage),
by = "cell"
) |>
dplyr::mutate(
lineage = dplyr::case_when(
lineage_ == "Blastocyst: Epiblast" ~ "EPI",
lineage_ == "Blastocyst: Hypoblast" ~ "HYP",
lineage_ %in% c(
"Blastocyst: Early Trophectoderm",
"Blastocyst: Trophectoderm"
) ~ "TE",
lineage_ %in% c(
"Inner Cell Mass"
) ~ "ICM",
lineage_ %in% c(
"Blastocyst: Inner Cell Mass-Trophectoderm Transition"
) ~ "ICM-TE",
# lineage_ %in% c("Other") ~ "NA",
TRUE ~ lineage
)
) |>
dplyr::select(!lineage_) |>
dplyr::mutate(
lineage = factor(
lineage,
levels = c(
"Pre-lineage",
"EPI", "HYP", "TE", "ICM", "ICM-TE"
)
)
)cells_blastocyst_LW119 <- scan(
file = file.path(
PROJECT_DIR, "genotyping",
"result_vartrix_LW121_no-duplicates_umi_mapq10_consensus",
"cells_LW119_blastocyst_snp20.txt"
),
what = "character"
)
cells_blastoid_LW119_LW121 <- embedding |>
dplyr::filter(
batch %in% c("LW119", "LW121"),
!cell %in% cells_blastocyst_LW119
) |>
dplyr::pull(cell)
cells_LW119_LW121 <- embedding |>
dplyr::filter(
batch %in% c("LW119", "LW121")
) |>
dplyr::pull(cell)
Map(
length,
list(cells_LW119_LW121, cells_blastoid_LW119_LW121, cells_blastocyst_LW119)
) |> unlist()[1] 7366 7165 201GEOM_POINT_STROKE <- 0.125color_palette_lineage_blastocyst <- c(
"Pre-lineage" = "#ffd8b1",
EPI = "#8ace7e",
HYP = "#ff684c",
TE = "#9467bd",
ICM = "#4E79A7",
"ICM-TE" = "#F97A1F",
"Non-blastocyst" = "grey70",
#
"Blastocyst; This study" = "#c969a1"
)
shape_palette_study <- c(
"This study" = 16,
"Petropoulos et al. 2016" = 15,
"Xiang et al. 2020" = 17,
"Yu et al. 2021" = 1,
"Yanagida et al. 2021" = 18,
"Zheng et al. 2019\nTyser et al. 2021" = 4,
"Other" = 4
)values <- embedding |>
dplyr::mutate(
value = lineage,
value = dplyr::case_when(
cell %in% cells_blastocyst_LW119 ~ "Blastocyst; This study",
batch %in% c("LW120", "LW122") ~ "Blastocyst; This study",
is.na(value) ~ "Non-blastocyst",
TRUE ~ as.character(value)
),
value = dplyr::case_when(
value %in% c("Blastocyst; This study") & (leiden %in% clusters_Pre_lineage) ~ "Pre-lineage",
value %in% c("Blastocyst; This study") & (leiden %in% clusters_ELCs) ~ "EPI",
value %in% c("Blastocyst; This study") & (leiden %in% clusters_HLCs) ~ "HYP",
value %in% c("Blastocyst; This study") & (leiden %in% clusters_TLCs) ~ "TE",
TRUE ~ as.character(value)
),
value = factor(
value,
levels = c(
"Non-blastocyst", levels(embedding$lineage)
)
)
) |>
dplyr::pull(value)
values_shape <- embedding |>
dplyr::mutate(
value = dplyr::case_when(
study %in% c(
"Tyser et al. 2021",
"Zheng et al. 2019"
) ~ "Zheng et al. 2019\nTyser et al. 2021", # "Other",
TRUE ~ as.character(study)
),
value = factor(
value,
levels = c(
"Petropoulos et al. 2016",
"Xiang et al. 2020",
"Yanagida et al. 2021",
"Yu et al. 2021",
"This study",
"Zheng et al. 2019\nTyser et al. 2021"
)
)
) |>
dplyr::pull(value)
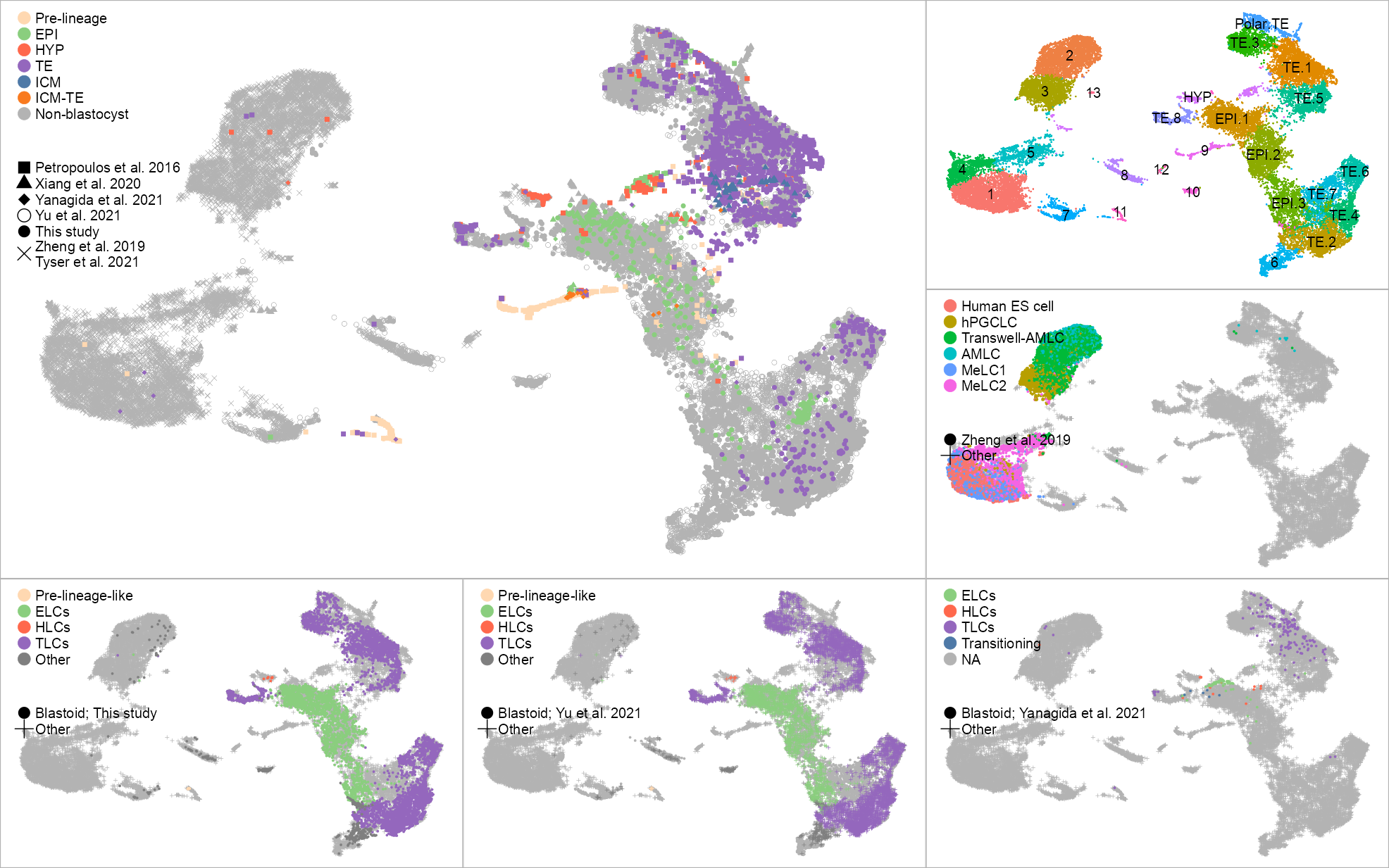
p_embedding_lineage_blastocyst <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
# label = glue::glue("{EMBEDDING_TITLE_PREFIX}; Blastocyst"),
color_labels = FALSE,
color_legend = TRUE,
shape = values_shape,
sort_values = TRUE,
shuffle_values = FALSE,
rasterise = TRUE,
geom_point_size = GEOM_POINT_SIZE * 1.5 * 2,
geom_point_stroke = 0.125,
geom_point_legend_ncol = 1
) +
ggplot2::guides(
shape = ggplot2::guide_legend(
override.aes = list(
size = 2,
alpha = 1,
stroke = c(0, 0, 0, 0.25, 0, 0.25)
),
ncol = 1,
order = 2
)
) +
ggplot2::scale_color_manual(
values = c(
color_palette_lineage_blastocyst
),
breaks = c(
levels(embedding$lineage),
"Blastocyst; This study",
"Non-blastocyst"
),
na.value = "grey70"
) +
ggplot2::scale_shape_manual(
values = shape_palette_study
) +
theme_customized_embedding(x = 0.035 / 2, y = 0.995, void = TRUE)cluster_labels <- list(
sort(unique(embedding$leiden)) |>
{
\(x) x[!x %in% c(clusters_ELCs, clusters_HLCs, clusters_TLCs)]
}() |>
tibble::enframe() |>
dplyr::mutate(
name = as.character(name)
),
sort(clusters_TLCs) |>
tibble::enframe() |>
dplyr::filter(value != 17) |>
dplyr::mutate(
name = 1:dplyr::n()
) |>
dplyr::mutate(
name = paste0("TE.", name)
),
sort(clusters_ELCs) |>
tibble::enframe() |>
dplyr::mutate(
name = paste0("EPI.", name)
),
data.frame(
name = "Polar.TE",
value = 17
),
sort(clusters_HLCs) |>
tibble::enframe() |>
dplyr::mutate(
name = paste0("HYP")
)
) |>
dplyr::bind_rows()
cluster_labels <- setNames(
object = cluster_labels$name,
nm = cluster_labels$value
)cluster_labels <- embedding |>
dplyr::group_by(leiden) |>
dplyr::summarise(
x = median(.data[[x_column]]),
y = median(.data[[y_column]])
) |>
dplyr::mutate(
label = cluster_labels[as.character(leiden)]
) |>
as.data.frame()p_embedding_leiden <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = embedding$leiden |> as.factor(),
# label = glue::glue("{EMBEDDING_TITLE_PREFIX}; Cluster"),
color_labels = FALSE,
color_legend = FALSE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE,
geom_point_stroke = GEOM_POINT_STROKE,
dpi = 900
) +
ggplot2::annotate(
geom = "text",
x = cluster_labels[, "x"],
y = cluster_labels[, "y"],
label = cluster_labels[, "label"],
family = "Arial",
color = "black",
size = 5 / ggplot2::.pt,
parse = TRUE
) +
theme_customized_embedding(void = TRUE)# Zheng et al. 2019; Amnion
p_embedding_lineage_amnion <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = embedding |>
dplyr::left_join(
cell_metadata_PRJNA555602,
by = "cell"
) |>
dplyr::select(
cell,
value = cell_type
) |>
dplyr::mutate(
value = forcats::fct_recode(
value,
"Human ES cell" = "Human_ES_cell"
)
) |>
dplyr::pull(value),
# label = glue::glue(),
color_labels = FALSE,
color_legend = TRUE,
shape = embedding |>
dplyr::left_join(
cell_metadata_PRJNA555602 |>
dplyr::select(cell, cell_type) |>
dplyr::mutate(
value = "1"
),
by = "cell"
) |>
dplyr::mutate(
value = dplyr::case_when(
is.na(value) ~ "Other",
TRUE ~ value
),
value = factor(
value,
)
) |>
dplyr::pull(value),
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = FALSE,
geom_point_size = GEOM_POINT_SIZE * 1.5,
geom_point_stroke = GEOM_POINT_STROKE
) +
ggplot2::scale_shape_manual(
values = c(16, 3),
labels = c("Zheng et al. 2019", "Other")
) +
ggplot2::scale_color_manual(
values = scales::hue_pal()(
n = nlevels(cell_metadata_PRJNA555602$cell_type)
),
breaks = forcats::fct_recode(
cell_metadata_PRJNA555602$cell_type,
"Human ES cell" = "Human_ES_cell"
) |> levels(),
na.value = "grey70"
) +
ggplot2::guides(
shape = ggplot2::guide_legend(
override.aes = list(
size = 2,
alpha = 1,
stroke = c(0, 0.25)
),
ncol = 1,
order = 2
)
) +
theme_customized_embedding(void = TRUE)color_palette_lineage_blastoid <- c(
"Pre-lineage-like" = "#ffd8b1",
ELCs = "#8ace7e",
HLCs = "#ff684c",
TLCs = "#9467bd",
Transitioning = "#4E79A7",
"Other" = "grey50"
)
# 16
clusters_Pre_lineage <- c(21, 23)
clusters_ELCs <- c(3, 6, 7)
clusters_HLCs <- c(20)
clusters_TLCs <- c(12, 14, 11, 2, 8, 17, 4, 10, 18)
values <- embedding |>
dplyr::mutate(
value = dplyr::case_when(
cell %in% cells_blastoid_LW119_LW121 &
leiden %in% clusters_Pre_lineage ~ "Pre-lineage-like",
#
cell %in% cells_blastoid_LW119_LW121 &
leiden %in% clusters_ELCs ~ "ELCs",
#
cell %in% cells_blastoid_LW119_LW121 &
leiden %in% clusters_HLCs ~ "HLCs",
#
cell %in% cells_blastoid_LW119_LW121 &
leiden %in% clusters_TLCs ~ "TLCs",
#
cell %in% cells_blastoid_LW119_LW121 & (
!leiden %in% c(
clusters_Pre_lineage,
clusters_ELCs,
clusters_HLCs,
clusters_TLCs
)
) ~ "Other"
),
value = factor(
value,
levels = c(
"Pre-lineage-like", "ELCs", "HLCs", "TLCs", "Other"
)
)
) |>
dplyr::pull(value)
p_embedding_lineage_blastocyst_like <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
# label = glue::glue("{EMBEDDING_TITLE_PREFIX}; Group"),
color_labels = FALSE,
color_legend = TRUE,
shape = embedding |>
dplyr::mutate(
value = dplyr::case_when(
cell %in% cells_blastoid_LW119_LW121 ~ "1",
TRUE ~ "Other"
),
value = factor(value)
) |>
dplyr::pull(value),
sort_values = TRUE,
shuffle_values = FALSE,
# rasterise = RASTERISED,
rasterise = FALSE,
geom_point_size = GEOM_POINT_SIZE * 1.5,
geom_point_stroke = 0.125,
) +
ggplot2::scale_color_manual(
values = color_palette_lineage_blastoid,
breaks = levels(values),
na.value = "grey70"
) +
theme_customized_embedding(void = TRUE) +
ggplot2::scale_shape_manual(
values = c(16, 3),
labels = c("Blastoid; This study", "Other")
) +
ggplot2::guides(
shape = ggplot2::guide_legend(
override.aes = list(
size = 2,
alpha = 1,
stroke = c(0, 0.25)
),
ncol = 1,
order = 2
)
)# Yu et al. 2021; PRJNA632839
p_embedding_blastoid_LW60_LW61 <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
# label = glue::glue(),
color_labels = FALSE,
color_legend = TRUE,
shape = embedding |>
dplyr::mutate(
value = dplyr::case_when(
batch %in% c("LW60", "LW61") ~ "1",
TRUE ~ "Other"
),
value = factor(value)
) |>
dplyr::pull(value),
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = FALSE,
geom_point_size = GEOM_POINT_SIZE * 1.5,
geom_point_stroke = GEOM_POINT_STROKE
) +
ggplot2::scale_color_manual(
values = c(color_palette_lineage_blastoid),
breaks = levels(values),
labels = levels(values),
na.value = "grey70"
) +
ggplot2::scale_shape_manual(
values = c(16, 3),
labels = c("Blastoid; Yu et al. 2021", "Other")
) +
ggplot2::guides(
shape = ggplot2::guide_legend(
override.aes = list(
size = 2,
alpha = 1,
stroke = c(0, 0.25)
),
ncol = 1,
order = 2
)
) +
theme_customized_embedding(void = TRUE)# Yanagida et al. 2021; PRJNA720968
p_embedding_lineage_PRJNA720968 <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = embedding |>
dplyr::left_join(
cell_metadata_PRJNA720968 |>
dplyr::filter(origin != "Blastocyst") |>
dplyr::select(cell, origin, lineage) |>
dplyr::mutate(
value = dplyr::case_when(
lineage %in% "Blastoid: Epiblast" ~ "ELCs",
lineage %in% "Blastoid: Hypoblast" ~ "HLCs",
lineage %in% "Blastoid: Trophectoderm" ~ "TLCs",
lineage %in% "Blastoid: Transitioning" ~ "Transitioning"
),
value = factor(
value,
levels = c(
"ELCs",
"HLCs",
"TLCs",
"Transitioning"
)
)
),
by = "cell"
) |>
dplyr::pull(value),
# label = glue::glue(),
color_labels = FALSE,
color_legend = TRUE,
shape = embedding |>
dplyr::left_join(
cell_metadata_PRJNA720968 |>
dplyr::filter(origin != "Blastocyst") |>
dplyr::select(cell, origin, lineage) |>
dplyr::mutate(
value = "1"
),
by = "cell"
) |>
dplyr::mutate(
value = dplyr::case_when(
is.na(value) ~ "Other",
TRUE ~ value
)
) |>
dplyr::pull(value),
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = FALSE,
geom_point_size = GEOM_POINT_SIZE * 1.5,
geom_point_stroke = GEOM_POINT_STROKE
) +
theme_customized_embedding(void = TRUE) +
ggplot2::scale_color_manual(
values = color_palette_lineage_blastoid,
na.value = "grey70"
) +
ggplot2::scale_shape_manual(
values = c(16, 3),
labels = c("Blastoid; Yanagida et al. 2021", "Other")
) +
ggplot2::guides(
shape = ggplot2::guide_legend(
override.aes = list(
size = 2,
alpha = 1,
stroke = c(0, 0.25)
),
ncol = 1,
order = 2
)
)layout <- "
AAB
AAC
DEF
"
list(
p_embedding_lineage_blastocyst,
p_embedding_leiden,
p_embedding_lineage_amnion,
p_embedding_lineage_blastocyst_like,
p_embedding_blastoid_LW60_LW61,
p_embedding_lineage_PRJNA720968
) |>
purrr::map(\(x) {
x + add_panel_border()
}) |>
purrr::reduce(`+`) +
patchwork::plot_layout(design = layout) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
) &
ggplot2::theme(
legend.background = ggplot2::element_blank(),
panel.background = ggplot2::element_blank(),
panel.grid.major = ggplot2::element_blank(),
panel.grid.minor = ggplot2::element_blank(),
#
# plot.background = ggplot2::element_blank(),
plot.background = element_rect(
colour = "grey70", fill = NA, linewidth = 0.25
),
#
axis.ticks.length = ggplot2::unit(0, "pt"),
panel.border = ggplot2::element_blank(),
plot.margin = ggplot2::unit(c(0, 0, 0, 0), "lines")
)
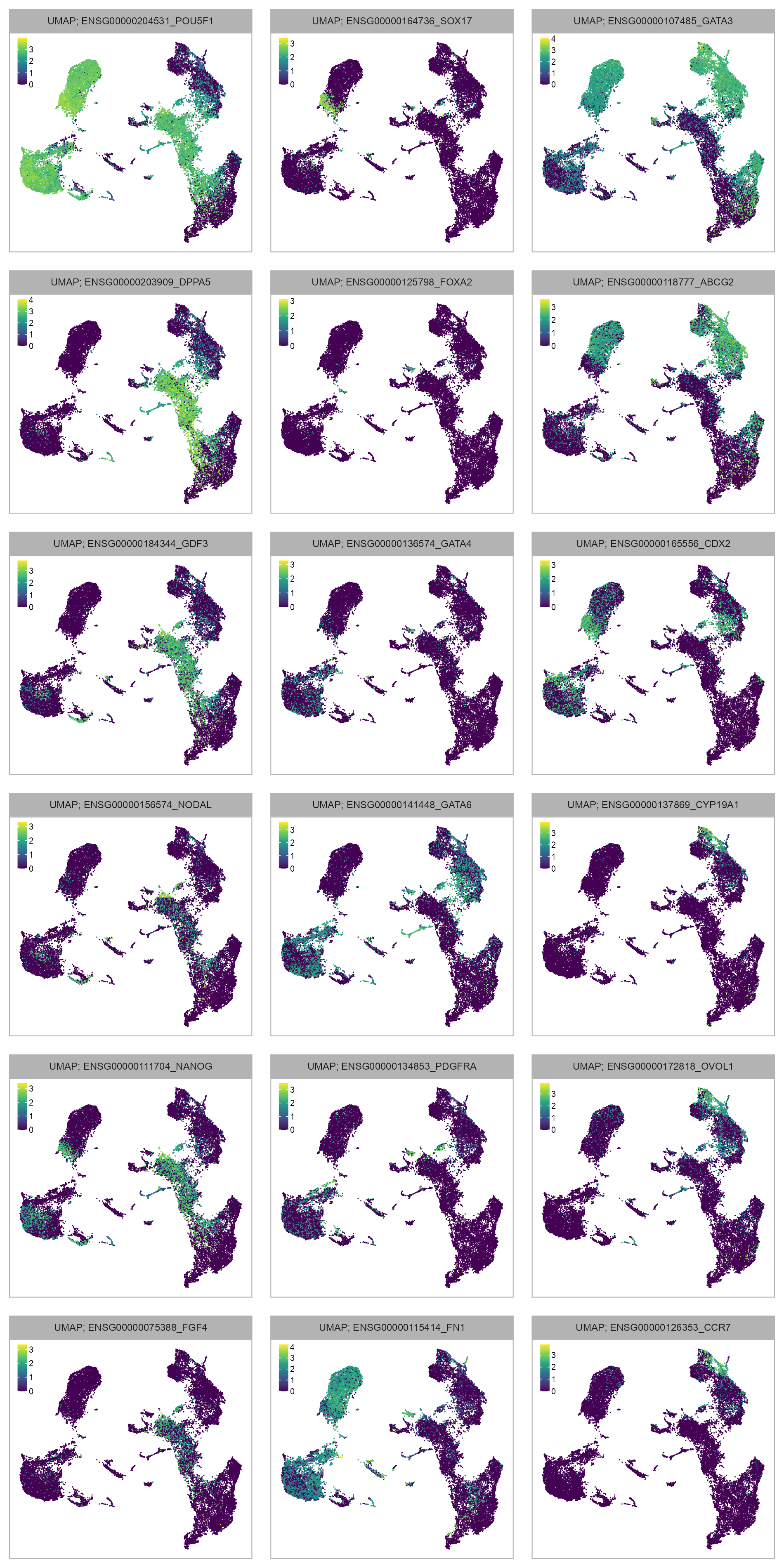
features_selected <- c(
"ENSG00000204531_POU5F1",
"ENSG00000203909_DPPA5",
"ENSG00000184344_GDF3",
"ENSG00000156574_NODAL",
"ENSG00000111704_NANOG",
"ENSG00000075388_FGF4",
#
"ENSG00000164736_SOX17",
"ENSG00000125798_FOXA2",
"ENSG00000136574_GATA4",
"ENSG00000141448_GATA6",
"ENSG00000134853_PDGFRA",
"ENSG00000115414_FN1",
#
"ENSG00000107485_GATA3",
"ENSG00000118777_ABCG2",
"ENSG00000165556_CDX2",
"ENSG00000137869_CYP19A1",
"ENSG00000172818_OVOL1",
"ENSG00000126353_CCR7"
)num_columns <- 3
purrr::map(features_selected, \(x) {
selected_feature <- x
values <- log10(calc_cpm(matrix_readcount_use)[x, embedding$cell] + 1)
# values[embedding$batch == "PRJEB11202"] <- NA
plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue("{EMBEDDING_TITLE_PREFIX}; {selected_feature}"),
color_labels = TRUE,
color_legend = TRUE,
sort_values = FALSE,
shuffle_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 1.5,
na_value = "grey70"
) +
theme_customized_embedding(
# x = CB_POSITION[1],
# y = CB_POSITION[2],
void = FALSE,
# legend_key_size = c(1.5, 1.5)
)
}) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = num_columns, byrow = FALSE) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
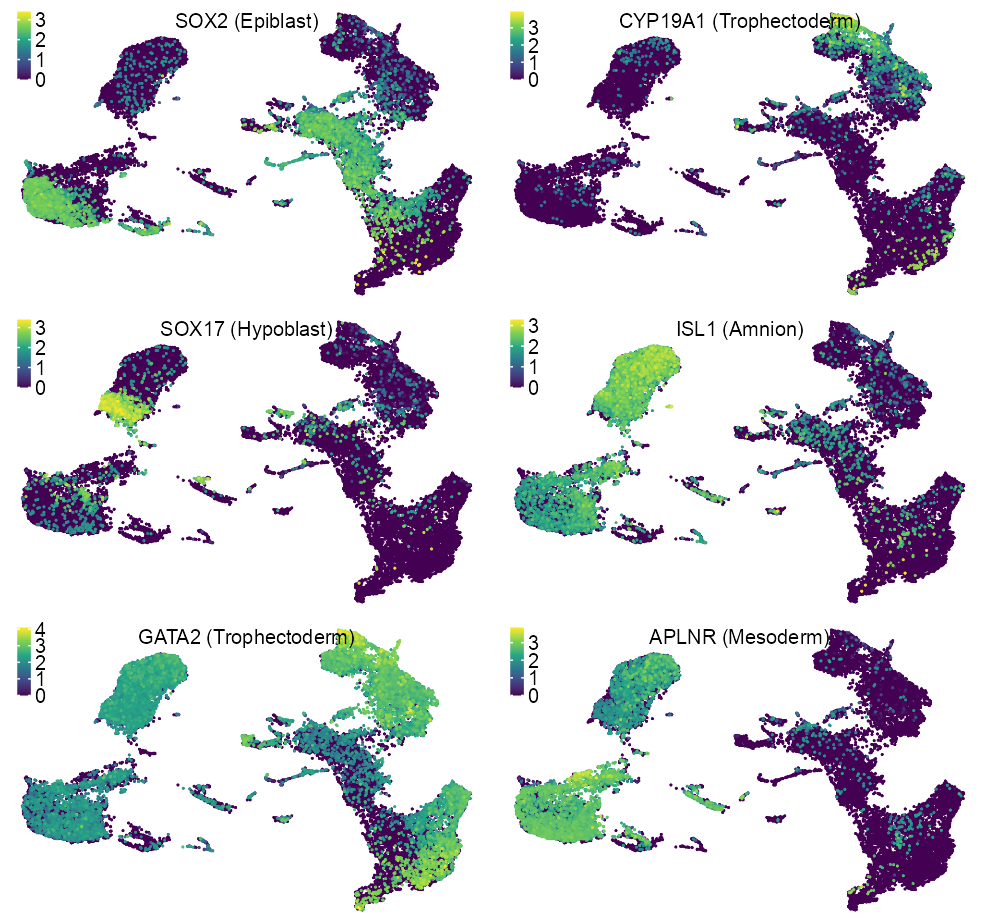
features_selected <- c(
"ENSG00000181449_SOX2",
# "ENSG00000187498_COL4A1",
"ENSG00000164736_SOX17",
"ENSG00000179348_GATA2",
"ENSG00000137869_CYP19A1",
#
"ENSG00000016082_ISL1",
# "ENSG00000094755_GABRP",
"ENSG00000134817_APLNR"
# "ENSG00000143320_CRABP2"
)
lineage_labels <- c(
"(Epiblast)",
"(Hypoblast)",
"(Trophectoderm)",
"(Trophectoderm)",
"(Amnion)",
"(Mesoderm)"
)x_label <- extract_ggplot2_axes_ranges(p_embedding_lineage_blastocyst)$x |>
unlist() |>
quantile(0.5)
y_label <- extract_ggplot2_axes_ranges(p_embedding_lineage_blastocyst)$y |>
unlist() |>
quantile(0.95)
purrr::map2(features_selected, lineage_labels, \(x, y) {
selected_feature <- x
values <- log10(
calc_cpm(matrix_readcount_use)[selected_feature, embedding$cell] + 1
)
# values[!embedding$cell %in% cells_blastoid_LW119_LW121] <- NA
plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
# label = glue::glue(y),
color_legend = TRUE,
sort_values = TRUE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 1.5,
na_value = "grey70"
) +
theme_customized_embedding(
# x = CB_POSITION[1],
# y = CB_POSITION[2],
void = TRUE,
legend_key_size = c(1.2, 1.2)
) +
ggplot2::annotate(
geom = "text",
x = x_label,
y = y_label,
label = stringr::str_c(
x |> stringr::str_remove(pattern = "^E.+_"),
y,
sep = " "
),
family = "Arial",
color = "black",
size = 5 / ggplot2::.pt,
hjust = 0.5,
vjust = 0
# parse = TRUE
)
}) |>
purrr::reduce(`+`) +
patchwork::plot_layout(nrow = 3, byrow = FALSE) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
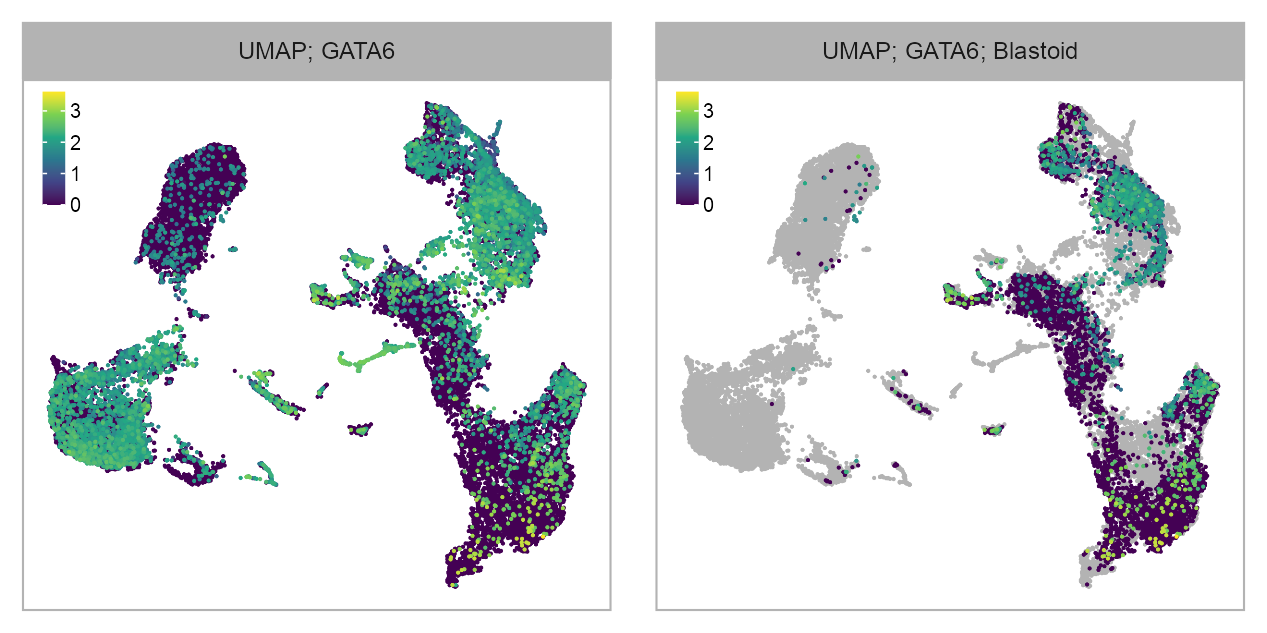
selected_feature <- "ENSG00000141448_GATA6"
values <- log10(
calc_cpm(matrix_readcount_use)[selected_feature, embedding$cell] + 1
)
p_embedding_GATA6 <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; ",
gsub("^E.+_", replacement = "", x = selected_feature)
),
color_labels = TRUE,
color_legend = TRUE,
sort_values = TRUE,
shuffle_values = FALSE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2,
na_value = "grey70"
) +
theme_customized_embedding()
values[!embedding$cell %in% cells_blastoid_LW119_LW121] <- NA
p_embedding_GATA6_blastoid <- plot_embedding(
data = embedding[, c(x_column, y_column)],
color = values,
label = glue::glue(
"{EMBEDDING_TITLE_PREFIX}; ",
gsub("^E.+_", replacement = "", x = selected_feature),
"; Blastoid"
),
color_labels = TRUE,
color_legend = TRUE,
sort_values = TRUE,
shuffle_values = FALSE,
rasterise = RASTERISED,
geom_point_size = GEOM_POINT_SIZE * 2,
na_value = "grey70"
) +
theme_customized_embedding()
list(
p_embedding_GATA6,
p_embedding_GATA6_blastoid
) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = 2, byrow = TRUE) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
) &
ggplot2::theme(
legend.background = ggplot2::element_blank(),
panel.background = ggplot2::element_blank(),
# panel.grid.major = ggplot2::element_blank(),
# panel.grid.minor = ggplot2::element_blank(),
#
plot.background = ggplot2::element_blank(),
# plot.background = element_rect(
# colour = "grey70", fill = NA, linewidth = 0.25
# ),
#
# panel.border = ggplot2::element_blank(),
# plot.margin = ggplot2::unit(c(0, 0, 0, 0), "lines")
)
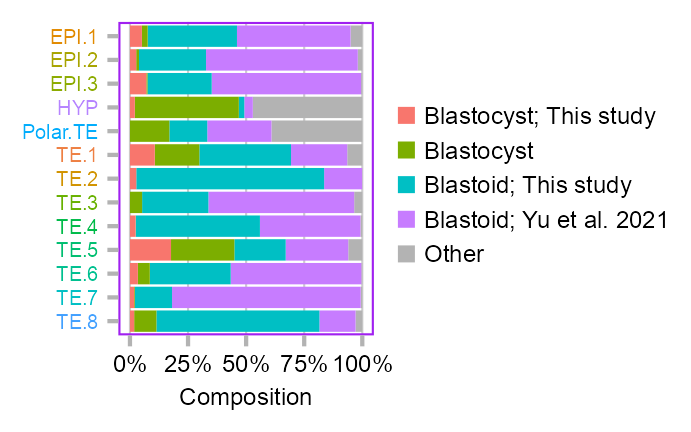
clusters_selected <- c(
clusters_ELCs,
clusters_HLCs,
clusters_TLCs
)
values <- embedding |>
dplyr::filter(
leiden %in% clusters_selected
) |>
dplyr::mutate(
value = dplyr::case_when(
cell %in% cells_blastocyst_LW119 ~ "Blastocyst; This study",
batch %in% c("LW120", "LW122") ~ "Blastocyst; This study",
cell %in% cells_blastoid_LW119_LW121 ~ "Blastoid; This study",
batch %in% c("LW60", "LW61") ~ "Blastoid; Yu et al. 2021",
lineage %in% c(
"EPI", "HYP", "ICM", "ICM-TE", "TE"
) ~ "Blastocyst",
TRUE ~ "Other"
),
value = factor(
value,
levels = c(
"Blastocyst; This study",
"Blastocyst",
"Blastoid; This study",
"Blastoid; Yu et al. 2021",
"Other"
) |> rev()
)
) |>
dplyr::pull(value)
values2 <- stringr::str_sort(cluster_labels[as.character(clusters_selected)],
numeric = TRUE
) |>
tibble::enframe() |>
dplyr::select(value) |>
dplyr::left_join(
cluster_labels |>
tibble::enframe() |>
dplyr::filter(name %in% clusters_selected)
)Joining with `by = join_by(value)`calc_group_composition(
data = embedding |>
dplyr::filter(
leiden %in% clusters_selected
) |>
dplyr::mutate(
leiden = factor(leiden, levels = rev(sort(unique(leiden)))),
value = values, # |> droplevels()
),
x = "leiden",
group = "value"
) |>
dplyr::mutate(
leiden = factor(leiden, levels = rev(values2$name))
) |>
plot_barplot(
x = "leiden",
y = "percentage",
z = "value",
legend_ncol = 1
) +
ggplot2::coord_flip() +
ggplot2::scale_fill_manual(
values = c(scales::hue_pal()(n = nlevels(values) - 1), "grey70"),
breaks = rev(levels(values))
) +
ggplot2::scale_x_discrete(
name = NULL,
labels = rev(values2$value)
) +
ggplot2::theme(
axis.text.y = ggplot2::element_text(
family = "Arial",
# color = "black",
color = setNames(
object = scales::hue_pal()(n = length(unique(embedding$leiden))),
nm = seq_len(length(unique(embedding$leiden)))
)[values2$name] |> rev(),
size = 5
),
#
panel.border = ggplot2::element_rect(
fill = NA, color = "purple"
),
strip.background = ggplot2::element_rect(
fill = "#b3b3b3", color = "purple"
),
)
clusters_selected_violin <- stringr::str_sort(
cluster_labels[as.character(clusters_selected_violin)],
numeric = TRUE
) |>
tibble::enframe() |>
dplyr::select(value) |>
dplyr::left_join(
cluster_labels |>
tibble::enframe()
) |>
dplyr::pull(name)
# blastoid
cells_violin <- purrr::map(clusters_selected_violin, \(x) {
embedding |>
dplyr::filter(
leiden == x,
cell %in% cells_blastoid_LW119_LW121
) |>
dplyr::pull(cell)
})
names(cells_violin) <- clusters_selected_violin
p_violin_blastoid <- plot_violin(
cells = cells_violin,
features = features_selected,
matrix_cpm = calc_cpm(matrix_readcount_use),
x_range_breaks = NULL
) +
ggplot2::scale_fill_manual(
name = NULL,
values = setNames(
object = scales::hue_pal()(n = length(unique(embedding$leiden))),
nm = seq_len(length(unique(embedding$leiden)))
)[as.character(clusters_selected_violin)]
) +
ggplot2::scale_color_manual(
name = NULL,
values = setNames(
object = scales::hue_pal()(n = length(unique(embedding$leiden))),
nm = seq_len(length(unique(embedding$leiden)))
)[as.character(clusters_selected_violin)]
) +
ggplot2::scale_y_discrete(
name = NULL,
labels = cluster_labels[clusters_selected_violin]
) +
ggplot2::labs(title = "Blastoids; This study; 10x Genomics") +
theme_customized_violin(
axis_title_size = 5,
axis_text_size = 5,
strip_text_size = 5,
strip_background_fill = "#d9d9d9ff",
panel_border_color = "purple"
) +
ggplot2::theme(
axis.text.y = ggplot2::element_text(
family = "Arial",
# color = "black",
color = setNames(
object = scales::hue_pal()(n = length(unique(embedding$leiden))),
nm = seq_len(length(unique(embedding$leiden)))
)[as.character(clusters_selected_violin)] |> rev(),
size = 5
),
#
plot.title = ggplot2::element_text(
family = "Arial",
size = 6,
margin = ggplot2::margin(
t = 0, r = 0, b = 1, l = 0,
unit = "mm"
),
hjust = 0.5
),
)p_violin_blastoid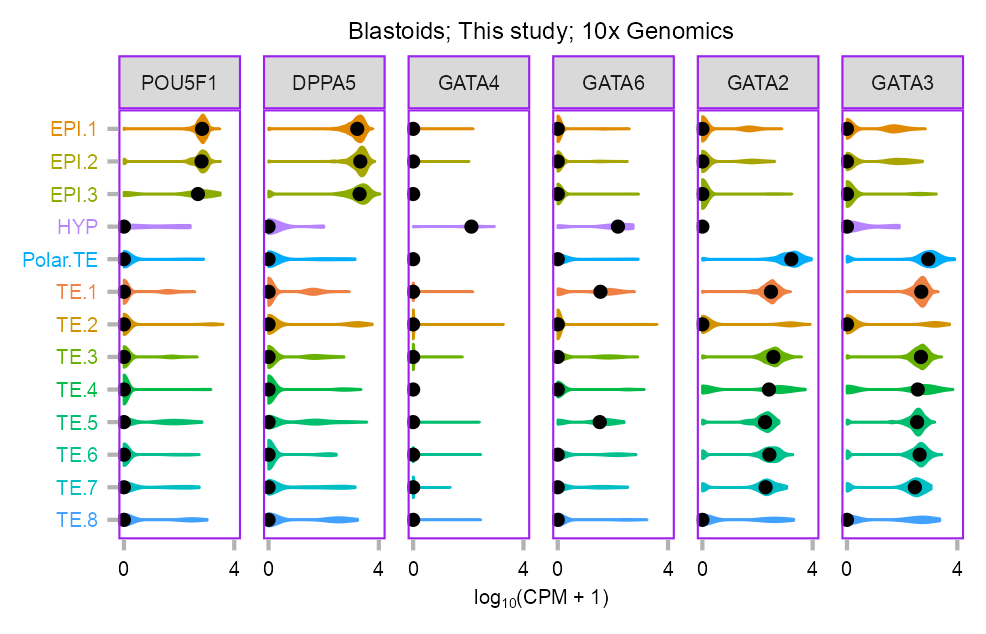
EDGER_METHOD <- "lrt"
LOG_FC_THRESHOLD <- 1de <- list(
ELCs = clusters_ELCs,
HLCs = clusters_HLCs,
TLCs = clusters_TLCs
) |>
purrr::map(\(x) {
cells_1 <- embedding |>
dplyr::filter(
leiden %in% x,
(
cell %in% c(cells_blastocyst_LW119)
) | batch %in% c("LW120", "LW122")
) |>
dplyr::pull(cell)
cells_2 <- embedding |>
dplyr::filter(
leiden %in% x,
!cell %in% c(cells_blastocyst_LW119) &
batch %in% c("LW119") | batch %in% c("LW121")
) |>
dplyr::pull(cell)
cat(x, "\n")
cat(length(cells_1), length(cells_2), "\n")
cat(sum(cells_1 %in% cells_2), "\n")
cat("=", "\n")
de <- run_pseudobulk_edgeR(
matrix = matrix_readcount_use,
cells_1 = cells_1,
cells_2 = cells_2,
method = EDGER_METHOD,
log_fc_threshold = LOG_FC_THRESHOLD
)
})3 6 7
264 1776
0
= number of pseudobulks: 8 (264) vs 10 (1776)20
8 9
0
= number of pseudobulks: 2 (10) vs 2 (10)Warning: Zero sample variances detected, have been offset away from zero12 14 11 2 8 17 4 10 18
635 5020
0
= number of pseudobulks: 10 (635) vs 9 (5020)de <- purrr::map(de, \(x) x[[3]])features_to_mark <- purrr::map(names(de), \(x) {
de[[x]] |>
dplyr::mutate(
category = x |>
stringr::str_remove(
pattern = "_.+$"
)
) |>
tibble::rownames_to_column(var = "feature") |>
dplyr::filter(
fdr < 0.05,
) |>
dplyr::mutate(
group = dplyr::case_when(
log_fc > 0 ~ "up",
TRUE ~ "down"
)
) |>
split(~group) |>
purrr::map(
\(xx) {
xx |>
dplyr::arrange(desc(abs(log_fc))) |>
dplyr::slice(1:5)
}
) |>
dplyr::bind_rows() |>
dplyr::mutate(
name = stringr::str_remove(string = feature, pattern = "_.+$"),
symbol = stringr::str_remove(string = feature, pattern = "^E.+_")
)
}) |>
dplyr::bind_rows()
p_scatter_de <- purrr::map(names(de), \(x) {
de[[x]] |>
dplyr::mutate(
category = x |>
stringr::str_remove(
pattern = "_.+$"
)
) |>
tibble::rownames_to_column(var = "feature")
}) |>
dplyr::bind_rows() |>
dplyr::mutate(
de = dplyr::case_when(
fdr < 0.05 & abs(log_fc) >= 2 ~ "1",
TRUE ~ "0"
)
) |>
ggplot2::ggplot(
aes(
x = log_cpm,
y = log_fc,
color = de
)
) +
ggrastr::rasterise(
input = ggplot2::geom_point(alpha = 0.8, size = 0.8, stroke = 0),
dpi = 900,
dev = "ragg_png"
) +
ggplot2::facet_grid(cols = vars(category), scales = "free")
p_scatter_de <- p_scatter_de +
ggplot2::scale_color_manual(values = c("grey70", "#00BFC4")) +
ggplot2::scale_x_continuous(
name = expression(paste("Average CPM ", "(log"[2], ")")),
) +
ggplot2::scale_y_continuous(
name = expression(paste("Fold change ", "(log"[2], ")")),
) +
ggplot2::ggtitle(label = "Blastocyst vs. blastoid") +
ggplot2::guides(color = "none") +
theme_customized2() +
ggplot2::theme(
axis.title.x = ggplot2::element_text(
family = "Arial",
size = 5,
),
axis.title.y = ggplot2::element_text(
family = "Arial",
size = 5,
margin = ggplot2::margin(t = 0, r = 0, b = 0, l = 0, unit = "mm")
),
axis.text = ggplot2::element_text(
family = "Arial",
color = "black",
size = 5
),
#
plot.title = ggplot2::element_text(
family = "Arial",
size = 6,
hjust = 0.5,
margin = ggplot2::margin(t = 0, r = 0, b = 1, l = 0, unit = "mm")
),
strip.text = ggplot2::element_text(
family = "Arial", size = 5
),
panel.border = ggplot2::element_rect(
fill = NA, color = "purple",
),
strip.background = ggplot2::element_rect(
fill = "#d9d9d9ff", color = "purple"
)
)g <- ggplot2::ggplot_gtable(
ggplot2::ggplot_build(p_scatter_de)
)
stript <- which(grepl("strip-t", g$layout$name))
k <- 1
for (i in stript) {
j <- which(grepl("rect", g$grobs[[i]]$grobs[[1]]$childrenOrder))
g$grobs[[i]]$grobs[[1]]$children[[j]]$gp$fill <- setNames(
object = c(ELCs = "#8ace7e", HLCs = "#ff684c", TLCs = "#9467bd"),
nm = seq_len(3)
)[k]
k <- k + 1
}
grid::grid.draw(g)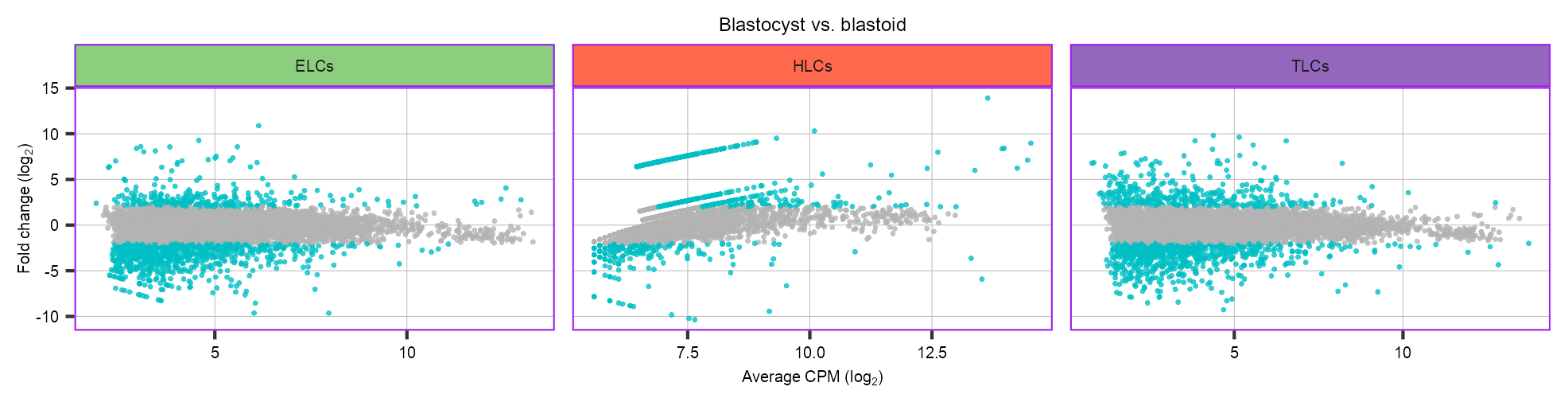
enriched_kegg <- tibble::tribble(
~id, ~description, ~GeneRatio, ~BgRatio, ~pvalue, ~category, ~group,
"hsa04390", "Hippo signaling pathway", "25/457", "157/7919", 3.04918e-06, "ELCs", "Blastocyst",
"hsa04350", "TGF-beta signaling pathway", "17/457", "97/7919", 3.34592e-05, "ELCs", "Blastocyst",
"hsa04520", "Adherens junction", "14/457", "71/7919", 4.29448e-05, "ELCs", "Blastocyst",
"hsa04550", "Signaling pathways regulating pluripotency of stem cells", "21/457", "143/7919", 6.51467e-05, "ELCs", "Blastocyst",
"hsa00190", "Oxidative phosphorylation", "17/457", "134/7919", 0.001733208, "ELCs", "Blastocyst",
"hsa00190", "Oxidative phosphorylation", "67/498", "134/7919", 1.43338e-45, "HLCs", "Blastocyst",
"hsa05208", "Chemical carcinogenesis - reactive oxygen species", "69/498", "221/7919", 5.58666e-31, "HLCs", "Blastocyst",
"hsa04723", "Retrograde endocannabinoid signaling", "31/498", "148/7919", 1.77784e-09, "HLCs", "Blastocyst",
"hsa04066", "HIF-1 signaling pathway", "13/498", "109/7919", 0.018816626, "HLCs", "Blastocyst",
"hsa00010", "Glycolysis / Gluconeogenesis", "8/498", "67/7919", 0.057839494, "HLCs", "Blastocyst",
"hsa04390", "Hippo signaling pathway", "20/493", "157/7919", 0.001693309, "TLCs", "Blastocyst",
"hsa04350", "TGF-beta signaling pathway", "14/493", "97/7919", 0.002566891, "TLCs", "Blastocyst",
"hsa00730", "Thiamine metabolism", "4/493", "15/7919", 0.011678143, "TLCs", "Blastocyst",
"hsa00561", "Glycerolipid metabolism", "9/493", "61/7919", 0.012499814, "TLCs", "Blastocyst",
"hsa04150", "mTOR signaling pathway", "17/493", "155/7919", 0.015861416, "TLCs", "Blastocyst",
"hsa04115", "p53 signaling pathway", "18/735", "73/6234", 0.001603023, "ELCs", "Blastoid",
"hsa04010", "MAPK signaling pathway", "47/735", "294/6234", 0.016867331, "ELCs", "Blastoid",
"hsa04926", "Relaxin signaling pathway", "23/735", "129/6234", 0.026882711, "ELCs", "Blastoid",
"hsa04928", "Parathyroid hormone synthesis, secretion and action", "19/735", "106/6234", 0.039701834, "ELCs", "Blastoid",
"hsa04152", "AMPK signaling pathway", "21/735", "121/6234", 0.043262071, "ELCs", "Blastoid",
"hsa04115", "p53 signaling pathway", "9/194", "73/6234", 0.000394004, "HLCs", "Blastoid",
"hsa04066", "HIF-1 signaling pathway", "10/194", "109/6234", 0.001979425, "HLCs", "Blastoid",
"hsa01521", "EGFR tyrosine kinase inhibitor resistance", "7/194", "79/6234", 0.011092936, "HLCs", "Blastoid",
"hsa04919", "Thyroid hormone signaling pathway", "9/194", "121/6234", 0.012770644, "HLCs", "Blastoid",
"hsa04010", "MAPK signaling pathway", "17/194", "294/6234", 0.009755139, "HLCs", "Blastoid",
"hsa04068", "FoxO signaling pathway", "28/611", "130/6234", 4.37072e-05, "TLCs", "Blastoid",
"hsa04115", "p53 signaling pathway", "17/611", "73/6234", 0.000531762, "TLCs", "Blastoid",
"hsa04066", "HIF-1 signaling pathway", "22/611", "109/6234", 0.000734062, "TLCs", "Blastoid",
"hsa05418", "Fluid shear stress and atherosclerosis", "25/611", "136/6234", 0.001379749, "TLCs", "Blastoid",
"hsa04926", "Relaxin signaling pathway", "24/611", "129/6234", 0.001439301, "TLCs", "Blastoid"
)purrr::map2(c("Blastocyst", "Blastoid"), c("#99B898", "#F9CDAD"), \(x, y) {
enriched_kegg |>
dplyr::select(
go_id = id,
term = description,
p_value = pvalue,
category,
group
) |>
dplyr::filter(group == x) |>
split(~category) |>
purrr::map(
\(x)
x |>
dplyr::arrange(p_value) |>
dplyr::slice(1:5) |>
dplyr::mutate(rank = 1:dplyr::n())
) |>
dplyr::bind_rows() |>
dplyr::mutate(
p_value_log = -log10(p_value)
) |>
{
\(z) {
z |>
ggplot2::ggplot(
ggplot2::aes(
x = -log10(p_value),
y = as.factor(rev(rank))
)
) +
ggplot2::geom_bar(
stat = "identity",
fill = y,
alpha = 0.8
) +
ggplot2::facet_grid(
cols = ggplot2::vars(category), scales = "free_x"
) +
ggplot2::scale_x_continuous(
name = glue::glue(
"KEGG; {x}; -log<sub>10</sub> p"
),
labels = scales::math_format(10^.x)
) +
ggplot2::guides(fill = "none") +
ggplot2::geom_text(
ggplot2::aes(
x = 0,
label = paste(" ", term, sep = ""),
group = NULL
),
size = 6 / ggplot2::.pt,
family = "Arial",
color = "black",
data = z,
hjust = 0
) +
# ggplot2::theme_classic()
ggplot2::theme(
axis.title.x = ggplot2::element_text(
family = "Arial", size = 5
),
axis.title.y = ggplot2::element_blank(),
axis.text.x = ggplot2::element_text(
family = "Arial", size = 5
),
axis.text.y = ggplot2::element_blank(),
axis.ticks.y = ggplot2::element_blank(),
axis.line.y = ggplot2::element_blank(),
legend.text = ggplot2::element_text(
family = "Arial", size = 5
),
legend.title = ggplot2::element_text(
family = "Arial", size = 5
)
) +
ggplot2::theme(
axis.line.x.bottom = element_line(
color = "purple", linewidth = 0.3
),
panel.background = ggplot2::element_blank(),
plot.background = ggplot2::element_blank(),
#
strip.background = ggplot2::element_rect(
fill = NA,
color = NA
),
#
strip.text = ggplot2::element_text(
family = "Arial",
size = 5,
color = "black",
hjust = 0.5,
margin = ggplot2::margin(
t = 0, r = 0, b = 1, l = 0,
unit = "mm"
)
),
axis.title.x = ggtext::element_markdown(),
)
}
}()
}) |>
purrr::reduce(`+`) +
patchwork::plot_layout(ncol = 1, byrow = FALSE) +
patchwork::plot_annotation(
theme = ggplot2::theme(plot.margin = ggplot2::margin())
)
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.3.1 (2023-06-16)
os macOS Ventura 13.5.2
system aarch64, darwin22.4.0
ui unknown
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz America/Chicago
date 2023-09-10
pandoc 2.19.2 @ /Users/jialei/.pyenv/shims/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
beeswarm 0.4.0 2021-06-01 [1] CRAN (R 4.3.0)
bit 4.0.5 2022-11-15 [1] CRAN (R 4.3.0)
bit64 4.0.5 2020-08-30 [1] CRAN (R 4.3.0)
cachem 1.0.8 2023-05-01 [1] CRAN (R 4.3.0)
callr 3.7.3 2022-11-02 [1] CRAN (R 4.3.0)
cli 3.6.1 2023-03-23 [1] CRAN (R 4.3.0)
colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.3.0)
commonmark 1.9.0 2023-03-17 [1] CRAN (R 4.3.0)
crayon 1.5.2 2022-09-29 [1] CRAN (R 4.3.0)
devtools 2.4.5.9000 2023-08-11 [1] Github (r-lib/devtools@163c3f2)
digest 0.6.33 2023-07-07 [1] CRAN (R 4.3.1)
dplyr * 1.1.3 2023-09-03 [1] CRAN (R 4.3.1)
ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.3.0)
evaluate 0.21 2023-05-05 [1] CRAN (R 4.3.0)
extrafont * 0.19 2023-01-18 [1] CRAN (R 4.3.0)
extrafontdb 1.0 2012-06-11 [1] CRAN (R 4.3.0)
fansi 1.0.4 2023-01-22 [1] CRAN (R 4.3.0)
farver 2.1.1 2022-07-06 [1] CRAN (R 4.3.0)
fastmap 1.1.1 2023-02-24 [1] CRAN (R 4.3.0)
forcats * 1.0.0.9000 2023-04-23 [1] Github (tidyverse/forcats@4a8525a)
fs 1.6.3 2023-07-20 [1] CRAN (R 4.3.1)
generics 0.1.3 2022-07-05 [1] CRAN (R 4.3.0)
ggbeeswarm 0.7.2 2023-04-29 [1] CRAN (R 4.3.0)
ggplot2 * 3.4.3.9000 2023-09-05 [1] Github (tidyverse/ggplot2@d180248)
ggrastr 1.0.2 2023-06-01 [1] CRAN (R 4.3.0)
ggtext 0.1.2 2022-09-16 [1] CRAN (R 4.3.0)
glue 1.6.2.9000 2023-04-23 [1] Github (tidyverse/glue@cbac82a)
gridtext 0.1.5 2022-09-16 [1] CRAN (R 4.3.0)
gt 0.9.0.9000 2023-09-02 [1] Github (rstudio/gt@c73eece)
gtable 0.3.4.9000 2023-08-22 [1] Github (r-lib/gtable@c410a54)
hms 1.1.3 2023-03-21 [1] CRAN (R 4.3.0)
htmltools 0.5.6 2023-08-10 [1] CRAN (R 4.3.1)
htmlwidgets 1.6.2 2023-03-17 [1] CRAN (R 4.3.0)
jsonlite 1.8.7 2023-06-29 [1] CRAN (R 4.3.1)
knitr 1.43 2023-05-25 [1] CRAN (R 4.3.0)
labeling 0.4.3 2023-08-29 [1] CRAN (R 4.3.1)
lattice 0.21-8 2023-04-05 [2] CRAN (R 4.3.1)
lifecycle 1.0.3 2022-10-07 [1] CRAN (R 4.3.0)
lubridate * 1.9.2.9000 2023-07-22 [1] Github (tidyverse/lubridate@cae67ea)
magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.3.0)
markdown 1.8 2023-08-23 [1] CRAN (R 4.3.1)
Matrix * 1.6-1 2023-08-14 [2] CRAN (R 4.3.1)
memoise 2.0.1 2021-11-26 [1] CRAN (R 4.3.0)
munsell 0.5.0 2018-06-12 [1] CRAN (R 4.3.0)
patchwork * 1.1.3.9000 2023-08-17 [1] Github (thomasp85/patchwork@51a6eff)
pillar 1.9.0 2023-03-22 [1] CRAN (R 4.3.0)
pkgbuild 1.4.2 2023-06-26 [1] CRAN (R 4.3.1)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.3.0)
pkgload 1.3.2.9000 2023-07-05 [1] Github (r-lib/pkgload@3cf9896)
png 0.1-8 2022-11-29 [1] CRAN (R 4.3.0)
prettyunits 1.1.1.9000 2023-04-23 [1] Github (r-lib/prettyunits@8706d89)
processx 3.8.2 2023-06-30 [1] CRAN (R 4.3.1)
ps 1.7.5 2023-04-18 [1] CRAN (R 4.3.0)
purrr * 1.0.2.9000 2023-08-11 [1] Github (tidyverse/purrr@ac4f5a9)
R.cache 0.16.0 2022-07-21 [1] CRAN (R 4.3.0)
R.methodsS3 1.8.2 2022-06-13 [1] CRAN (R 4.3.0)
R.oo 1.25.0 2022-06-12 [1] CRAN (R 4.3.0)
R.utils 2.12.2 2022-11-11 [1] CRAN (R 4.3.0)
R6 2.5.1.9000 2023-04-23 [1] Github (r-lib/R6@e97cca7)
ragg 1.2.5 2023-01-12 [1] CRAN (R 4.3.0)
Rcpp 1.0.11 2023-07-06 [1] CRAN (R 4.3.1)
readr * 2.1.4.9000 2023-08-03 [1] Github (tidyverse/readr@80e4dc1)
remotes 2.4.2.9000 2023-06-09 [1] Github (r-lib/remotes@8875171)
reticulate 1.31 2023-08-10 [1] CRAN (R 4.3.1)
rlang 1.1.1.9000 2023-06-09 [1] Github (r-lib/rlang@c55f602)
rmarkdown 2.24.2 2023-09-07 [1] Github (rstudio/rmarkdown@8d2d9b8)
rstudioapi 0.15.0.9000 2023-09-07 [1] Github (rstudio/rstudioapi@19c80c0)
Rttf2pt1 1.3.12 2023-01-22 [1] CRAN (R 4.3.0)
sass 0.4.7 2023-07-15 [1] CRAN (R 4.3.1)
scales 1.2.1 2022-08-20 [1] CRAN (R 4.3.0)
sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.3.0)
stringi 1.7.12 2023-01-11 [1] CRAN (R 4.3.0)
stringr * 1.5.0.9000 2023-08-11 [1] Github (tidyverse/stringr@08ff36f)
styler * 1.10.2 2023-08-30 [1] Github (r-lib/styler@1976817)
systemfonts 1.0.4 2022-02-11 [1] CRAN (R 4.3.0)
textshaping 0.3.6 2021-10-13 [1] CRAN (R 4.3.0)
tibble * 3.2.1.9005 2023-05-28 [1] Github (tidyverse/tibble@4de5c15)
tidyr * 1.3.0.9000 2023-04-23 [1] Github (tidyverse/tidyr@0764e65)
tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.3.0)
tidyverse * 2.0.0.9000 2023-04-23 [1] Github (tidyverse/tidyverse@8ec2e1f)
timechange 0.2.0 2023-01-11 [1] CRAN (R 4.3.0)
tzdb 0.4.0 2023-05-12 [1] CRAN (R 4.3.0)
usethis 2.2.2.9000 2023-07-11 [1] Github (r-lib/usethis@467ff57)
utf8 1.2.3 2023-01-31 [1] CRAN (R 4.3.0)
vctrs 0.6.3 2023-06-14 [1] CRAN (R 4.3.0)
vipor 0.4.5 2017-03-22 [1] CRAN (R 4.3.0)
viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.3.0)
vroom 1.6.3.9000 2023-04-30 [1] Github (tidyverse/vroom@89b6aac)
withr 2.5.0 2022-03-03 [1] CRAN (R 4.3.0)
xfun 0.40 2023-08-09 [1] CRAN (R 4.3.1)
xml2 1.3.5 2023-07-06 [1] CRAN (R 4.3.1)
yaml 2.3.7 2023-01-23 [1] CRAN (R 4.3.0)
[1] /opt/homebrew/lib/R/4.3/site-library
[2] /opt/homebrew/Cellar/r/4.3.1/lib/R/library
─ Python configuration ───────────────────────────────────────────────────────
python: /Users/jialei/.pyenv/shims/python
libpython: /Users/jialei/.pyenv/versions/mambaforge-22.9.0-3/lib/libpython3.10.dylib
pythonhome: /Users/jialei/.pyenv/versions/mambaforge-22.9.0-3:/Users/jialei/.pyenv/versions/mambaforge-22.9.0-3
version: 3.10.9 | packaged by conda-forge | (main, Feb 2 2023, 20:26:08) [Clang 14.0.6 ]
numpy: /Users/jialei/.pyenv/versions/mambaforge-22.9.0-3/lib/python3.10/site-packages/numpy
numpy_version: 1.24.3
numpy: /Users/jialei/.pyenv/versions/mambaforge-22.9.0-3/lib/python3.10/site-packages/numpy
NOTE: Python version was forced by RETICULATE_PYTHON
──────────────────────────────────────────────────────────────────────────────Styling 1 files:
interpret_blastoids_public.qmd ✔
────────────────────────────────────────
Status Count Legend
✔ 1 File unchanged.
ℹ 0 File changed.
✖ 0 Styling threw an error.
────────────────────────────────────────