PHAGE-ATAC; Anti-CD8 Phage Hashing Single-cell ATAC-seq Using CD8 T Cells from Four Human Donors¶
Dataset: PHAGE-ATAC: four anti-CD8 phage hashtags and a subsequent hashing experiment using CD8 T cells from four human donors
Fiskin, E., Lareau, C.A., Ludwig, L.S., Eraslan, G., Liu, F., Ring, A.M., Xavier, R.J., and Regev, A. (2021). Single-cell profiling of proteins and chromatin accessibility using PHAGE-ATAC. Nat. Biotechnol. 40, 374–381.
Preparation¶
Download fastq files from Gene Expression Omnibus.
Inspect fastq files (This is a phage-derived tag library built on top of the single-cell ATAC-seq library, we will need all 3 end reads).
$ zcat SRR12588752_1.fastq.gz | head
@SRR12588752.1 NB501583:726:HMCKKBGXF:1:11101:16677:1031 length=34
CAGCTNTTCCTGCGCTCGACACCGTTACTTGTGT
+SRR12588752.1 NB501583:726:HMCKKBGXF:1:11101:16677:1031 length=34
AAAAA#EEEEEEEEEEEEEEEEEEEEEEEEEEEE
@SRR12588752.2 NB501583:726:HMCKKBGXF:1:11101:14655:1033 length=34
CAGCTNTTCCTGCGCTGCTTACCGTAACTTGTGT
+SRR12588752.2 NB501583:726:HMCKKBGXF:1:11101:14655:1033 length=34
AAAAA#EEEEEEEAEEEEEEEEEEEEEEEEEEEE
@SRR12588752.3 NB501583:726:HMCKKBGXF:1:11101:17414:1034 length=34
CAGCTNTTCCTGCGCTCGACACCGTTACTTGTGT
$ zcat SRR12588752_2.fastq.gz | head
@SRR12588752.1 NB501583:726:HMCKKBGXF:1:11101:16677:1031 length=16
NNNNAGNNNNNATGNN
+SRR12588752.1 NB501583:726:HMCKKBGXF:1:11101:16677:1031 length=16
####AE#####EEE##
@SRR12588752.2 NB501583:726:HMCKKBGXF:1:11101:14655:1033 length=16
NNAACTNNNNNAAAAN
+SRR12588752.2 NB501583:726:HMCKKBGXF:1:11101:14655:1033 length=16
##//A6#####////#
@SRR12588752.3 NB501583:726:HMCKKBGXF:1:11101:17414:1034 length=16
NNTCAGTNNNTCAGGN
$ zcat SRR12588752_3.fastq.gz | head
@SRR12588752.1 NB501583:726:HMCKKBGXF:1:11101:16677:1031 length=34
GATACCGNGGNNNNNNNNNNNNNNNNNNNNNNNN
+SRR12588752.1 NB501583:726:HMCKKBGXF:1:11101:16677:1031 length=34
AAAAAEE#EE########################
@SRR12588752.2 NB501583:726:HMCKKBGXF:1:11101:14655:1033 length=34
GATACCGCGGTGTNTNANNNCNNNNNAGNNNCNN
+SRR12588752.2 NB501583:726:HMCKKBGXF:1:11101:14655:1033 length=34
AAAAAEEEEEEEE#E#E###E#####EE###E##
@SRR12588752.3 NB501583:726:HMCKKBGXF:1:11101:17414:1034 length=34
GATACCGCGGTGTATNANNNGNNNNNAANNNGNN
Prepare cell barcodes (downloaded from the manuscript’s GitHub repository). These are the cell-associated barcodes, they are generated based on the single-cell ATAC-seq library.
$ wget https://raw.githubusercontent.com/evgenijfiskin/phage-atac/master/cd8_hashing/data/barcodes.tsv
Inspect cell barcodes.
$ wc -l barcodes.tsv
8366
$ head barcodes.tsv
AAACGAAAGACCATAA-1
AAACGAAAGGAGGCGA-1
AAACGAAAGGCGATTG-1
AAACGAACAAGAGATT-1
AAACGAACACAGTTAC-1
AAACGAACACTGATTG-1
AAACGAACACTTACAG-1
AAACGAACAGCAGGTA-1
AAACGAACAGCGTACC-1
AAACGAAGTTGCAGAG-1
Prepare feature barcodes. CDR3 barcode sequences (phage-derived tags, PDTs) can be found in Supplementary Table 4 and are truncated to only keep the variable parts.
CD8Nb PH-A |
GATACCGCGGTGTATTATTGCGCAAAGGACGCGG |
CD8Nb PH-B |
GATACCGCGGTGTATTATTGCGCTAAAGACGCGG |
CD8Nb PH-C |
CAGCTCTTCCTGCGCTGCTTACCGTAACTTGTGT |
CD8Nb PH-D |
CAGCTCTTCCTGCGCTGCTTACAGTGACCTGTGT |
$ cat feature_barcodes_R3_truncated.tsv
CD8Nb_PH-A CAAAGGACGCGG
CD8Nb_PH-B CTAAAGACGCGG
$ cat feature_barcodes_R1_truncated.tsv
CD8Nb_PH-C CGTAACTTGTGT
CD8Nb_PH-D AGTGACCTGTGT
First, we screened reads for the constant sequence
(GATACCGCGGTGTATTATTGCG) at the beginning of the CDR3 barcode
sequences on read 3 using cutadapt (version 3.7).
$ cutadapt \
--cores 0 \
--front GATACCGCGGTGTATTATTGCG \
--minimum-length 12:16 \
--trimmed-only \
--output SRR12588752_3_trimmed.fq.gz --paired-output SRR12588752_2_trimmed.fq.gz \
SRR12588752_3.fastq.gz SRR12588752_2.fastq.gz
Preview the filtering result: 51,140,637 out of 54,274,791 (94.2%) read pairs are kept for phage-derived tag (PDT) identification.
== Read fate breakdown ==
Pairs that were too short: 652,917 (1.2%)
Pairs discarded as untrimmed: 2,481,237 (4.6%)
Pairs written (passing filters): 51,140,637 (94.2%)
Then, for read 1 (CAGCTCTTCCTGCGCTGCTTAC).
$ cutadapt \
--cores 0 \
--front CAGCTCTTCCTGCGCTGCTTAC \
--minimum-length 12:16 \
--trimmed-only \
--output SRR12588752_1_trimmed.fq.gz --paired-output SRR12588752_2_trimmed.fq.gz \
SRR12588752_1.fastq.gz SRR12588752_2.fastq.gz
Preview the filtering result: 25,988,762 out of 54,274,791 (47.9%) read pairs are kept for phage-derived tag (PDT) identification.
== Read fate breakdown ==
Pairs that were too short: 16,644 (0.0%)
Pairs discarded as untrimmed: 28,269,385 (52.1%)
Pairs written (passing filters): 25,988,762 (47.9%)
QC¶
The first 10,000 read pairs are sampled (set by -n, default
100,000) for quality control. The -t option can be used to set
the number of threads. By default, diagnostic results and plots are
generated in the qc directory (set by --output_directory), and
the full length of read 1 and read 2 are searched against reference cell
and feature barcodes, respectively. The per base content of both read
pairs and the distribution of matched barcode positions are summarized.
Use -r1_c and/or -r2_c to limit the search range, and -cb_n
and/or -fb_n to set the mismatch tolerance for cell and/or feature
barcode matching (default 3).
This library was constructed using 10x Genomics’ Chromium Single Cell
ATAC Reagent Kits, where the 16-base pair cell barcode is sequenced
during the i5 index read. In Cell Ranger ATAC, the raw 16 bp
sequences may be transformed into their reverse-complement counterparts
as cell barcodes in the outputs. To achieve the same result in fba,
use -cb_rc to reverse-complement the cell barcode sequences.
To achieve the same result in fba,
R3¶
$ fba qc \
-1 SRR12588752_2_trimmed.fq.gz \
-2 SRR12588752_3_trimmed.fq.gz \
-w barcodes.tsv \
-f feature_barcodes_R3_truncated.txt \
-cb_rc \
-n 10000
This library is constructed using the Chromium Single Cell ATAC Reagent Kits and sequenced on Illumina NextSeq 500. The GC content of cell barcodes (read 2) are quite even.
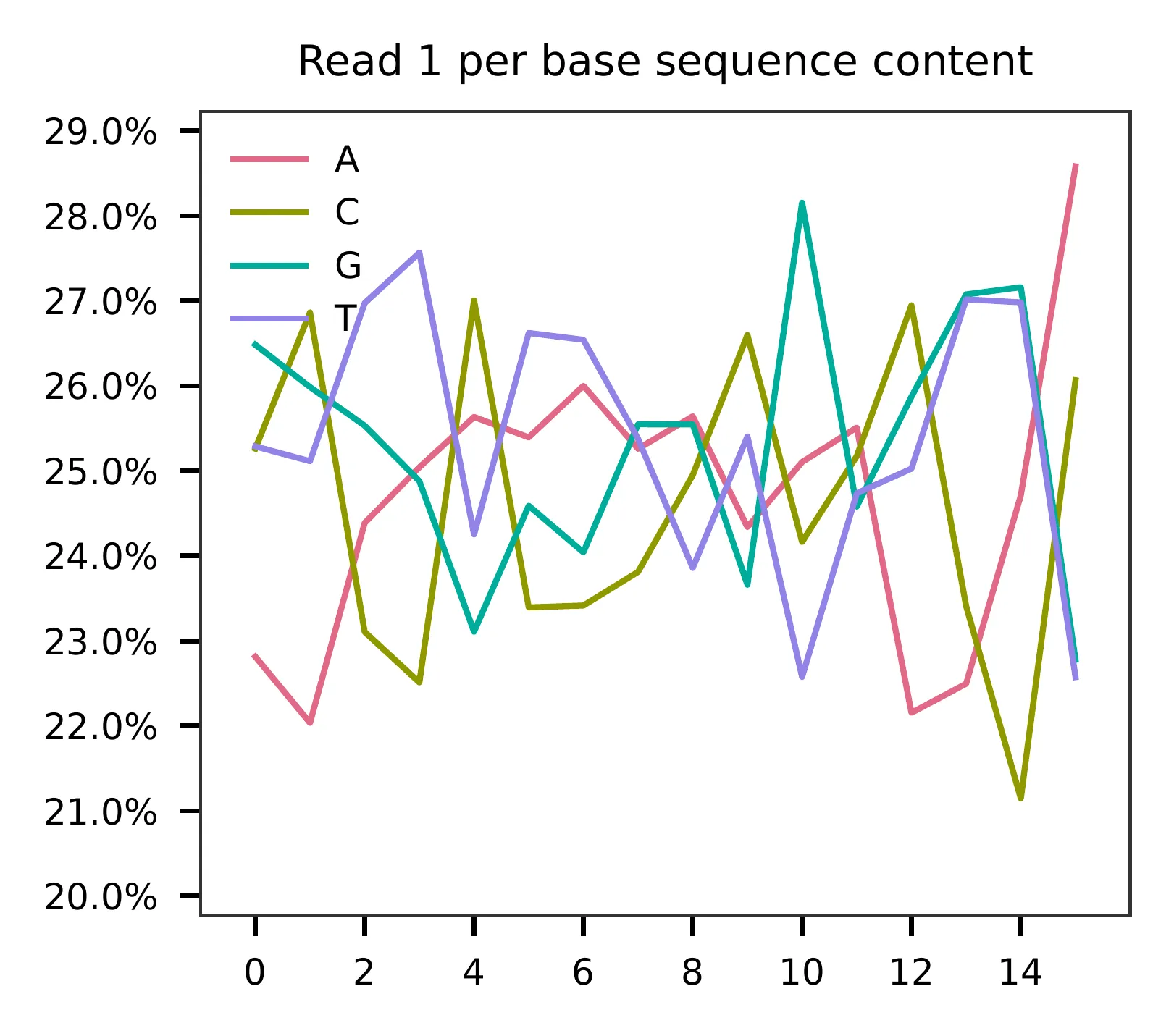

As for read 3, based on the per base content, it suggests low complexity.

The detailed qc results are stored in the
feature_barcoding_output.tsv.gz file. The matching_pos columns
indicate the matched positions on reads, while the
matching_description columns indicate mismatches in the format of
substitutions:insertions:deletions.
$ gzip -dc feature_barcoding_output.tsv.gz | head
read1_seq cell_barcode cb_num_mismatches read2_seq feature_barcode fb_num_mismatches
NTGTTGCTGGTTAGAA CTGTTGCTGGTTAGAA 1 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
NTCGACCGATTGCGTA GTCGACCGATTGCGTA 1 CTAAAGACGCGG CD8Nb_PH-B_CTAAAGACGCGG 0
GCCGAACTGTTAGAAG GCCGAACTGTTAGAAG 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
TGAGCGCACACCTTGA TGAGCGCACACCTTGA 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
AATTCTGCTTGGCTGC AATTCTGCTTGGCTGC 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
GGAATGGTGACCGTGC GGAATGGTGACCGTGC 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
AGGAATTGATTCGCCT AGGAATTGATTCGCCT 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
CCAAGTTGATAATAGG CCAAGTTGATAATAGG 0 CTAAAGACGCGG CD8Nb_PH-B_CTAAAGACGCGG 0
CCGCAAGTGAATCCAC CCGCAAGTGAATCCAC 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
R1¶
$ fba qc \
-1 SRR12588752_2_trimmed.fq.gz \
-2 SRR12588752_1_trimmed.fq.gz \
-w barcodes.tsv \
-f feature_barcodes_R1_truncated.txt \
-cb_rc \
-n 10000
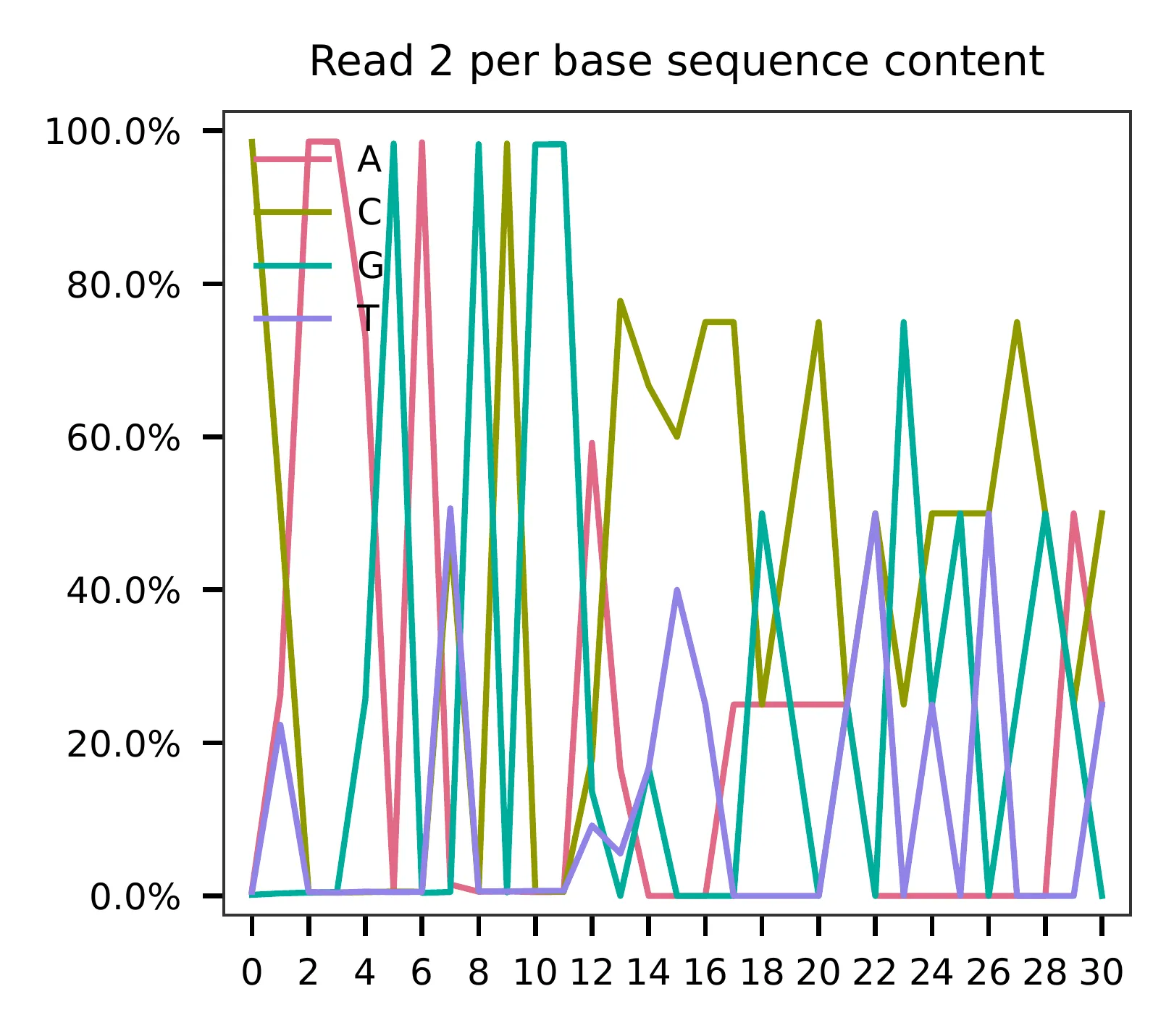
For read 1, the per base content plot suggests low complexity, with a high proportion of constant bases at the beginning of the reads.
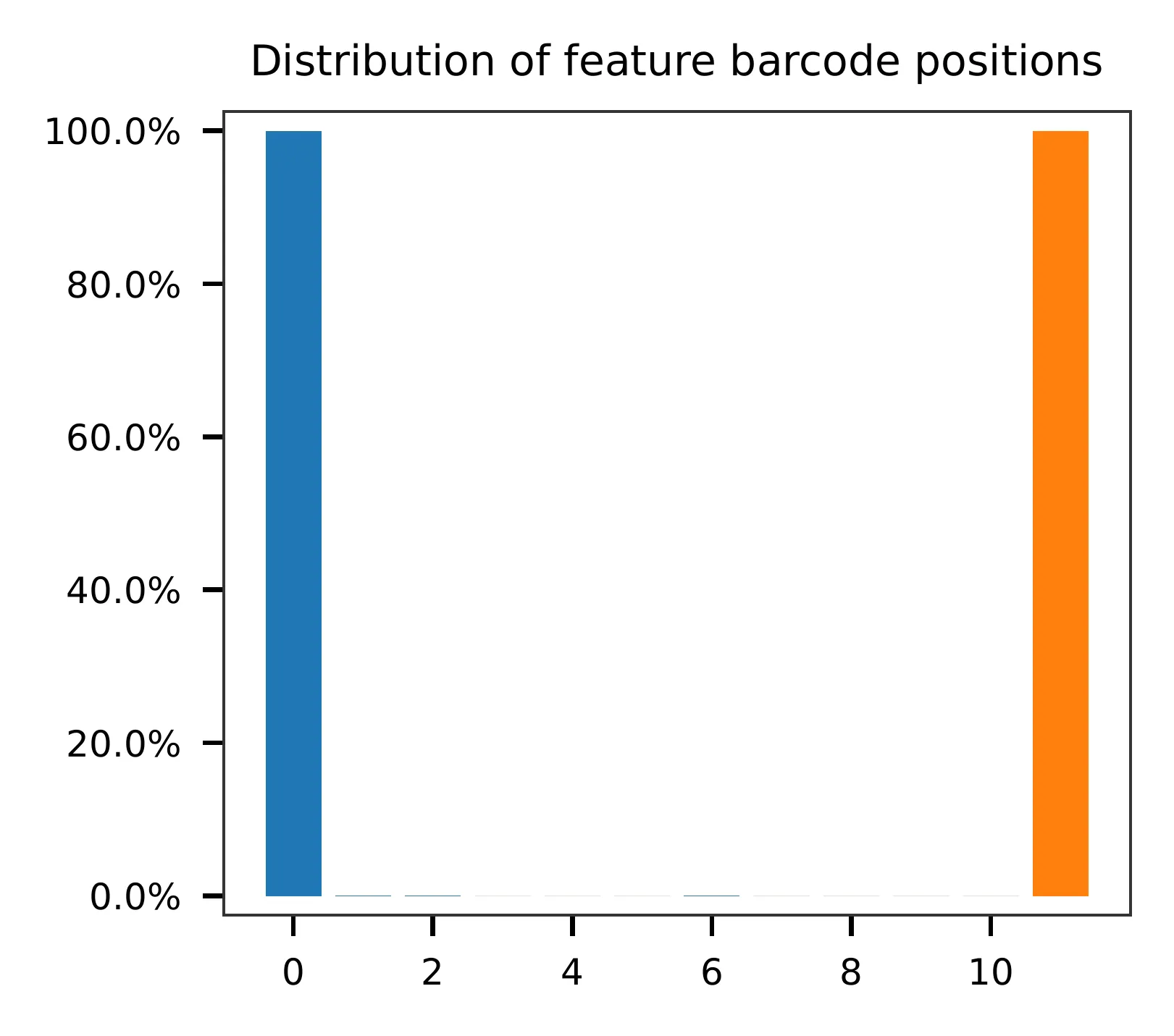
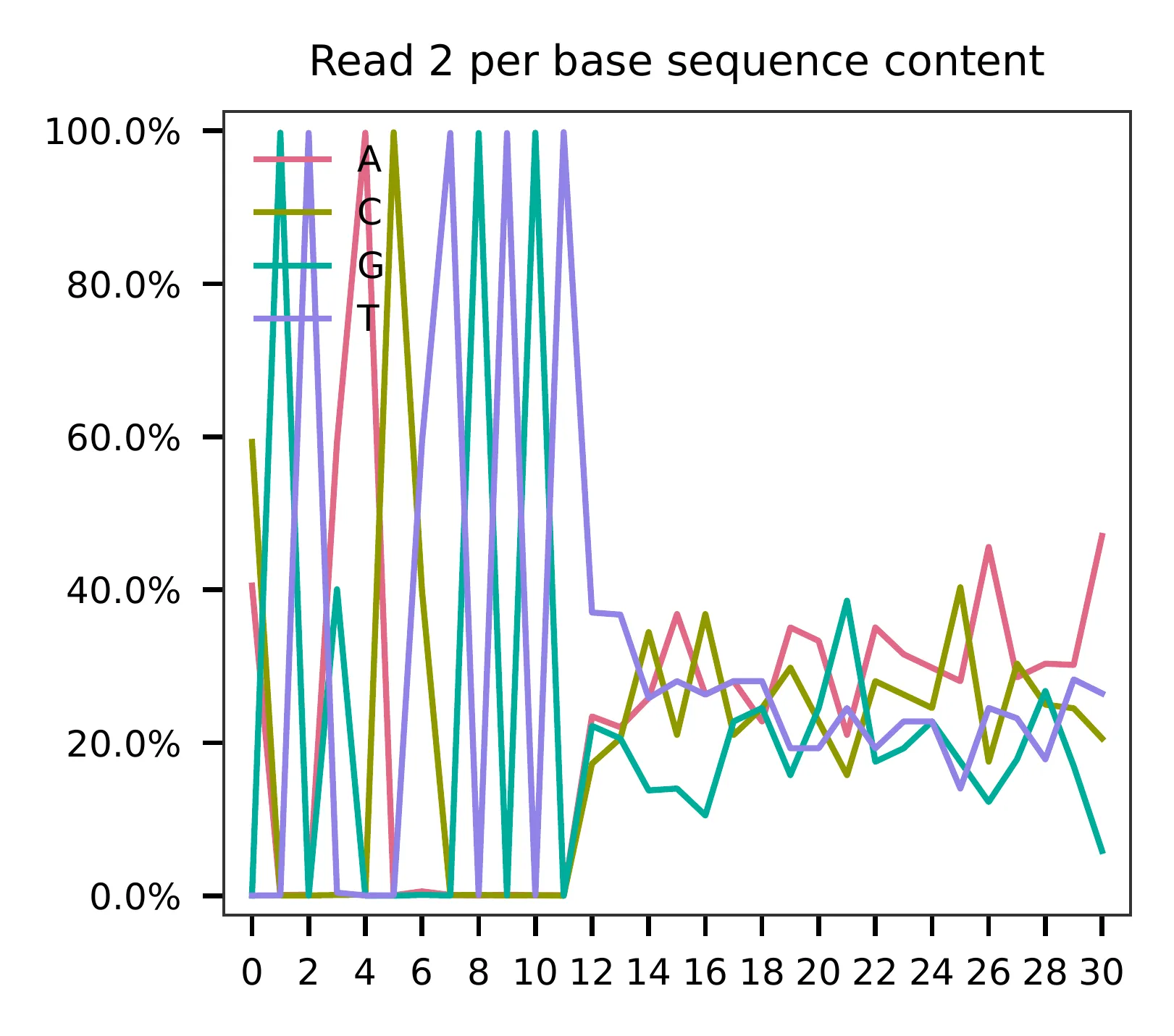
The detailed qc results are stored in the
feature_barcoding_output.tsv.gz file. The matching_pos columns
indicate the matched positions on reads, while the
matching_description columns indicate mismatches in the format of
substitutions:insertions:deletions.
$ gzip -dc feature_barcoding_output.tsv.gz | head
read1_seq cell_barcode cb_num_mismatches read2_seq feature_barcode fb_num_mismatches
NCTCGGGACGTCTGGC ACTCGGGACGTCTGGC 1 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
NCTAAGACTTTATGGC GCTAAGACTTTATGGC 1 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
NACGGAAGATCGTAAC CACGGAAGATCGTAAC 1 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
NTGTTGTGAGTCCCGA GTGTTGTGAGTCCCGA 1 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
CCTCCTGCTATCAGGG CCTCCTGCTATCAGGG 0 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
GTTGATTCTCGAAGCA GTTGATTCTCGAAGCA 0 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
TGGTTAGACTCCGTAA TGGTTAGACTCCGTAA 0 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
GCCTCTTGACTGGGTC GCCTCTTGACTGGGTC 0 CGTAACTTGTGT CD8Nb_PH-C_CGTAACTTGTGT 0
AGGTAGCGAGAGTAAT AGGTAGCGAGAGTAAT 0 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
Barcode extraction¶
R3¶
Search ranges are set to 0,16 on read 2 and 0,12 on read 3. One
mismatch for cell and feature barcodes (-cb_m, -cf_m) are
allowed. Use -cb_rc to reverse-complement cell barcode sequences in
the output if needed.
$ fba extract \
-1 SRR12588752_2_trimmed.fq.gz \
-2 SRR12588752_3_trimmed.fq.gz \
-w barcodes.tsv \
-f feature_barcodes_R3_truncated.txt \
-o feature_barcoding_output_R3.tsv.gz \
-r1_c 0,16 \
-r2_c 0,12 \
-cb_m 1 \
-fb_m 1 \
-cb_rc
Preview of result.
$ gzip -dc feature_barcoding_output_R3.tsv.gz | head
read1_seq cell_barcode cb_num_mismatches read2_seq feature_barcode fb_num_mismatches
NTGTTGCTGGTTAGAA CTGTTGCTGGTTAGAA 1 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
NTCGACCGATTGCGTA GTCGACCGATTGCGTA 1 CTAAAGACGCGG CD8Nb_PH-B_CTAAAGACGCGG 0
GCCGAACTGTTAGAAG GCCGAACTGTTAGAAG 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
TGAGCGCACACCTTGA TGAGCGCACACCTTGA 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
AATTCTGCTTGGCTGC AATTCTGCTTGGCTGC 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
GGAATGGTGACCGTGC GGAATGGTGACCGTGC 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
AGGAATTGATTCGCCT AGGAATTGATTCGCCT 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
CCAAGTTGATAATAGG CCAAGTTGATAATAGG 0 CTAAAGACGCGG CD8Nb_PH-B_CTAAAGACGCGG 0
CCGCAAGTGAATCCAC CCGCAAGTGAATCCAC 0 CAAAGGACGCGG CD8Nb_PH-A_CAAAGGACGCGG 0
Result summary.
10,543,901 out of 51,140,637 read pairs have valid cell and feature barcodes.
2022-03-13 00:13:02,564 - fba.__main__ - INFO - fba version: 0.0.12
2022-03-13 00:13:02,564 - fba.__main__ - INFO - Initiating logging ...
2022-03-13 00:13:02,564 - fba.__main__ - INFO - Python version: 3.10
2022-03-13 00:13:02,564 - fba.__main__ - INFO - Using extract subcommand ...
2022-03-13 00:13:02,589 - fba.levenshtein - INFO - Number of reference cell barcodes: 8,366
2022-03-13 00:13:02,590 - fba.levenshtein - INFO - Number of reference feature barcodes: 2
2022-03-13 00:13:02,590 - fba.levenshtein - INFO - Read 1 coordinates to search: [0, 16)
2022-03-13 00:13:02,590 - fba.levenshtein - INFO - Read 2 coordinates to search: [0, 12)
2022-03-13 00:13:02,590 - fba.levenshtein - INFO - Cell barcode maximum number of mismatches: 1
2022-03-13 00:13:02,590 - fba.levenshtein - INFO - Feature barcode maximum number of mismatches: 1
2022-03-13 00:13:02,590 - fba.levenshtein - INFO - Read 1 maximum number of N allowed: 3
2022-03-13 00:13:02,590 - fba.levenshtein - INFO - Read 2 maximum number of N allowed: 3
2022-03-13 00:13:02,809 - fba.levenshtein - INFO - Matching ...
2022-03-13 00:16:00,978 - fba.levenshtein - INFO - Read pairs processed: 10,000,000
2022-03-13 00:18:58,488 - fba.levenshtein - INFO - Read pairs processed: 20,000,000
2022-03-13 00:21:55,956 - fba.levenshtein - INFO - Read pairs processed: 30,000,000
2022-03-13 00:24:53,698 - fba.levenshtein - INFO - Read pairs processed: 40,000,000
2022-03-13 00:27:51,819 - fba.levenshtein - INFO - Read pairs processed: 50,000,000
2022-03-13 00:28:12,045 - fba.levenshtein - INFO - Number of read pairs processed: 51,140,637
2022-03-13 00:28:12,045 - fba.levenshtein - INFO - Number of read pairs w/ valid barcodes: 10,543,901
2022-03-13 00:28:12,060 - fba.__main__ - INFO - Done.
R1¶
Search ranges are set to 0,16 on read 2 and 0,12 on read 1. One
mismatch for cell and feature barcodes (-cb_m, -cf_m) are
allowed. Use -cb_rc to reverse-complement cell barcode sequences in
the output if needed.
$ fba extract \
-1 SRR12588752_2_trimmed.fq.gz \
-2 SRR12588752_1_trimmed.fq.gz \
-w barcodes.tsv \
-f feature_barcodes_R1_truncated.txt \
-o feature_barcoding_output_R1.tsv.gz \
-r1_c 0,16 \
-r2_c 0,12 \
-cb_m 1 \
-fb_m 1 \
-cb_rc
Preview of result.
$ gzip -dc feature_barcoding_output_R1.tsv.gz | head
read1_seq cell_barcode cb_num_mismatches read2_seq feature_barcode fb_num_mismatches
NCTCGGGACGTCTGGC ACTCGGGACGTCTGGC 1 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
NCTAAGACTTTATGGC GCTAAGACTTTATGGC 1 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
NACGGAAGATCGTAAC CACGGAAGATCGTAAC 1 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
NTGTTGTGAGTCCCGA GTGTTGTGAGTCCCGA 1 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
CCTCCTGCTATCAGGG CCTCCTGCTATCAGGG 0 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
GTTGATTCTCGAAGCA GTTGATTCTCGAAGCA 0 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
TGGTTAGACTCCGTAA TGGTTAGACTCCGTAA 0 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
GCCTCTTGACTGGGTC GCCTCTTGACTGGGTC 0 CGTAACTTGTGT CD8Nb_PH-C_CGTAACTTGTGT 0
AGGTAGCGAGAGTAAT AGGTAGCGAGAGTAAT 0 AGTGACCTGTGT CD8Nb_PH-D_AGTGACCTGTGT 0
Result summary.
11,128,546 out of 25,988,762 read pairs have valid cell and feature barcodes.
2022-03-12 23:29:33,460 - fba.__main__ - INFO - fba version: 0.0.12
2022-03-12 23:29:33,460 - fba.__main__ - INFO - Initiating logging ...
2022-03-12 23:29:33,460 - fba.__main__ - INFO - Python version: 3.10
2022-03-12 23:29:33,460 - fba.__main__ - INFO - Using extract subcommand ...
2022-03-12 23:29:33,488 - fba.levenshtein - INFO - Number of reference cell barcodes: 8,366
2022-03-12 23:29:33,488 - fba.levenshtein - INFO - Number of reference feature barcodes: 2
2022-03-12 23:29:33,488 - fba.levenshtein - INFO - Read 1 coordinates to search: [0, 16)
2022-03-12 23:29:33,488 - fba.levenshtein - INFO - Read 2 coordinates to search: [0, 12)
2022-03-12 23:29:33,488 - fba.levenshtein - INFO - Cell barcode maximum number of mismatches: 1
2022-03-12 23:29:33,488 - fba.levenshtein - INFO - Feature barcode maximum number of mismatches: 1
2022-03-12 23:29:33,488 - fba.levenshtein - INFO - Read 1 maximum number of N allowed: 3
2022-03-12 23:29:33,488 - fba.levenshtein - INFO - Read 2 maximum number of N allowed: 3
2022-03-12 23:29:33,707 - fba.levenshtein - INFO - Matching ...
2022-03-12 23:33:10,471 - fba.levenshtein - INFO - Read pairs processed: 10,000,000
2022-03-12 23:36:47,019 - fba.levenshtein - INFO - Read pairs processed: 20,000,000
2022-03-12 23:38:56,544 - fba.levenshtein - INFO - Number of read pairs processed: 25,988,762
2022-03-12 23:38:56,544 - fba.levenshtein - INFO - Number of read pairs w/ valid barcodes: 11,128,546
2022-03-12 23:38:56,558 - fba.__main__ - INFO - Done.
Matrix generation¶
Only fragments with correctly matched cell and feature barcodes are
included. Use -ul to set the UMI length (default 12). Setting to
0 means no UMIs and read counts are summarized instead. Use
-cb_rc to reverse-complement cell barcode sequences in the output
matrix if needed. The generated feature count matrix can be easily
imported into well-established single cell analysis packages such as
Seurat and Scanpy.
$ fba count \
-i feature_barcoding_output_R1.tsv.gz \
-i feature_barcoding_output_R3.tsv.gz \
-o matrix_featurecount.csv.gz \
-ul 0
Result summary.
39.9 % (21,672,447 out of 54,274,791) of total read pairs have valid cell and feature barcodes. The median number of reads per cell for this phage-derived tag library is 2,261.0.
2022-03-13 00:36:01,502 - fba.__main__ - INFO - fba version: 0.0.12
2022-03-13 00:36:01,502 - fba.__main__ - INFO - Initiating logging ...
2022-03-13 00:36:01,502 - fba.__main__ - INFO - Python version: 3.9
2022-03-13 00:36:01,502 - fba.__main__ - INFO - Using count subcommand ...
2022-03-13 00:36:02,348 - fba.count - INFO - UMI-tools version: 1.1.1
2022-03-13 00:36:02,348 - fba.count - INFO - UMI length set to 0, ignoring UMI information. Skipping arguments: "-us/--umi_start".
2022-03-13 00:36:02,348 - fba.count - INFO - Header: read1_seq cell_barcode cb_num_mismatches read2_seq feature_barcode fb_num_mismatches
2022-03-13 00:36:20,914 - fba.count - INFO - Number of read pairs processed: 21,672,447
2022-03-13 00:36:20,917 - fba.count - INFO - Number of cell barcodes detected: 8,366
2022-03-13 00:36:20,917 - fba.count - INFO - Number of features detected: 4
2022-03-13 00:36:20,917 - fba.count - INFO - Counting ...
2022-03-13 00:36:21,009 - fba.count - INFO - Total reads: 21,672,447
2022-03-13 00:36:21,016 - fba.count - INFO - Median number of reads per cell: 2,261.0
2022-03-13 00:36:21,103 - fba.__main__ - INFO - Done.
Demultiplexing¶
Negative binomial distribution¶
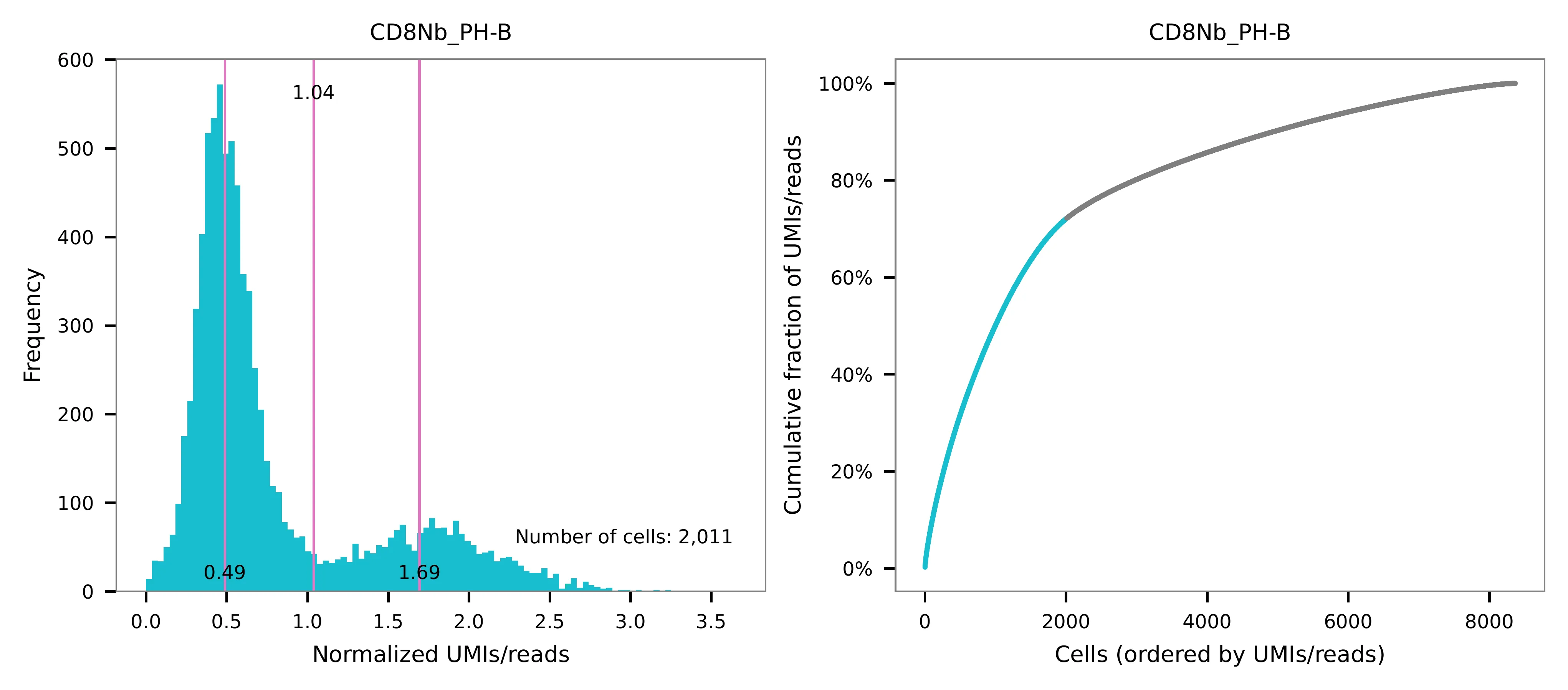
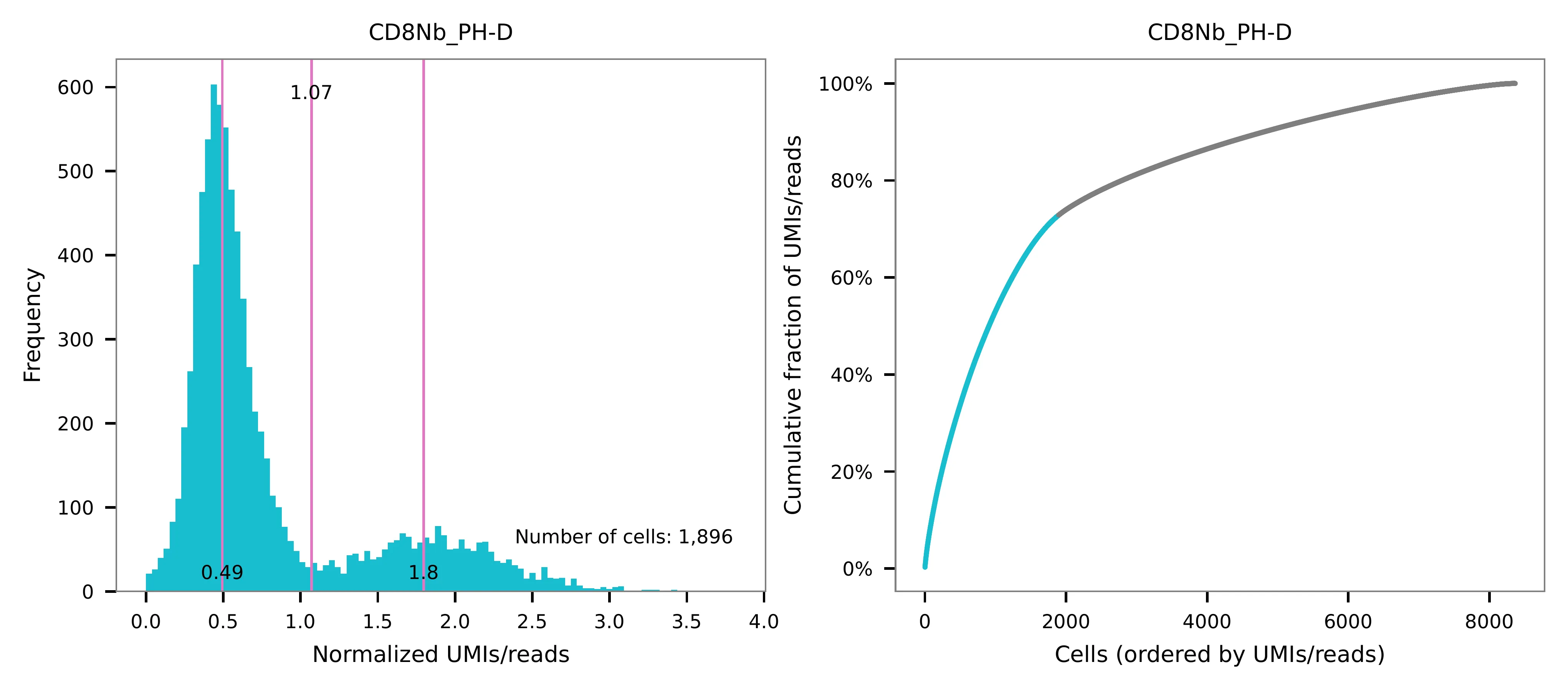
Cells are demultiplexed based on the feature count matrix using
demultiplexing method 1 (set by -dm), which is implemented based
on the method described by Stoeckius, M., et al. (2018) with some
modifications. The output directory for demultiplexing is set by
--output_directory (default demultiplexed). A cell identity
matrix is generated, where 0 indicates negative and 1 indicates
positive. To adjust the quantile threshold for demultiplexing, use
-q (default 0.9999). To generate visualization plots, set
-v.
$ fba demultiplex \
-i matrix_featurecount.csv.gz \
-q 0.99 \
-v
2022-03-13 00:47:41,569 - fba.__main__ - INFO - fba version: 0.0.12
2022-03-13 00:47:41,569 - fba.__main__ - INFO - Initiating logging ...
2022-03-13 00:47:41,569 - fba.__main__ - INFO - Python version: 3.10
2022-03-13 00:47:41,569 - fba.__main__ - INFO - Using demultiplex subcommand ...
2022-03-13 00:47:49,145 - fba.__main__ - INFO - Skipping arguments: "-p/--prob"
2022-03-13 00:47:49,145 - fba.demultiplex - INFO - Output directory: demultiplexed
2022-03-13 00:47:49,145 - fba.demultiplex - INFO - Demultiplexing method: 1
2022-03-13 00:47:49,146 - fba.demultiplex - INFO - UMI normalization method: clr
2022-03-13 00:47:49,146 - fba.demultiplex - INFO - Visualization: On
2022-03-13 00:47:49,146 - fba.demultiplex - INFO - Visualization method: tsne
2022-03-13 00:47:49,146 - fba.demultiplex - INFO - Loading feature count matrix: matrix_featurecount.csv.gz ...
2022-03-13 00:47:49,324 - fba.demultiplex - INFO - Number of cells: 8,366
2022-03-13 00:47:49,324 - fba.demultiplex - INFO - Number of positive cells for a feature to be included: 200
2022-03-13 00:47:49,327 - fba.demultiplex - INFO - Number of features: 4 / 4 (after filtering / original in the matrix)
2022-03-13 00:47:49,327 - fba.demultiplex - INFO - Features: CD8Nb_PH-A CD8Nb_PH-B CD8Nb_PH-C CD8Nb_PH-D
2022-03-13 00:47:49,327 - fba.demultiplex - INFO - Total UMIs/reads: 21,672,447 / 21,672,447
2022-03-13 00:47:49,328 - fba.demultiplex - INFO - Median number of UMIs/reads per cell: 2,261.0 / 2,261.0
2022-03-13 00:47:49,328 - fba.demultiplex - INFO - Demultiplexing ...
2022-03-13 00:48:53,685 - fba.demultiplex - INFO - Generating heatmap ...
2022-03-13 00:48:59,759 - fba.demultiplex - INFO - Embedding ...
2022-03-13 00:49:20,128 - fba.__main__ - INFO - Done.
Heatmap of the relative abundance of features (phage-derived tags, PDTs) across all cells. Each column represents a single cell. This is a re-creation of Fig. 3b in Fiskin, E., et al. (2021).
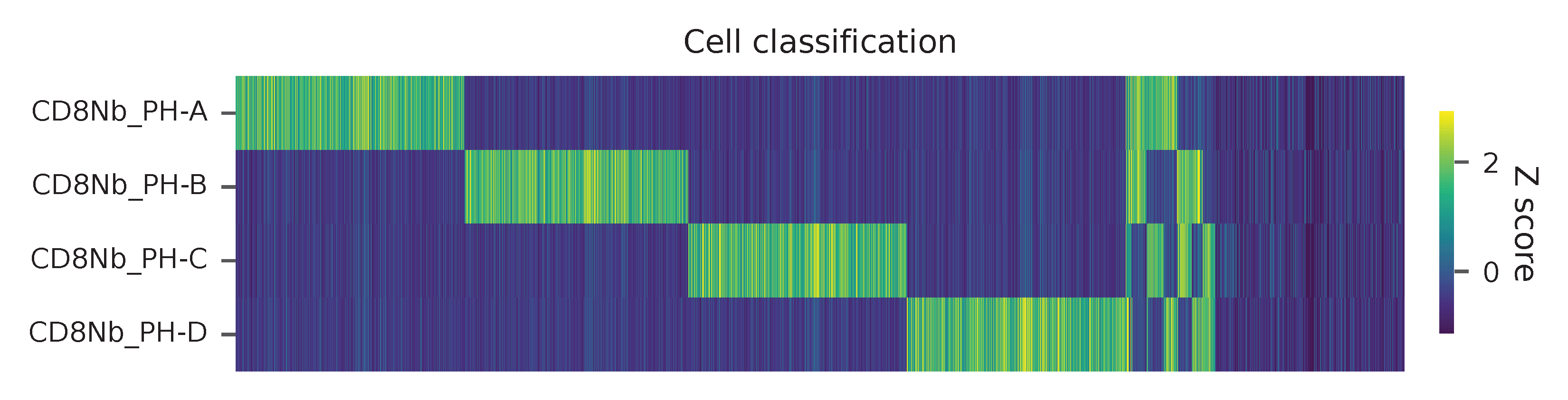
Preview the demultiplexing result: the numbers of singlets, multiplets and negatives are 6,373 (76.2%), 639 (7.6%), and 1,354 (16.2%), respectively.
In [1]: import pandas as pd
In [2]: m = pd.read_csv("demultiplexed/matrix_cell_identity.csv.gz", index_col=0)
In [3]: m.loc[:, m.sum(axis=0) == 1].sum(axis=1)
Out[3]:
CD8Nb_PH-A 1640
CD8Nb_PH-B 1601
CD8Nb_PH-C 1564
CD8Nb_PH-D 1568
dtype: int64
In [4]: sum(m.sum(axis=0) == 1)
Out[4]: 6373
In [5]: sum(m.sum(axis=0) > 1)
Out[5]: 639
In [6]: sum(m.sum(axis=0) == 0)
Out[6]: 1354
In [7]: m.shape
Out[7]: (4, 8366)
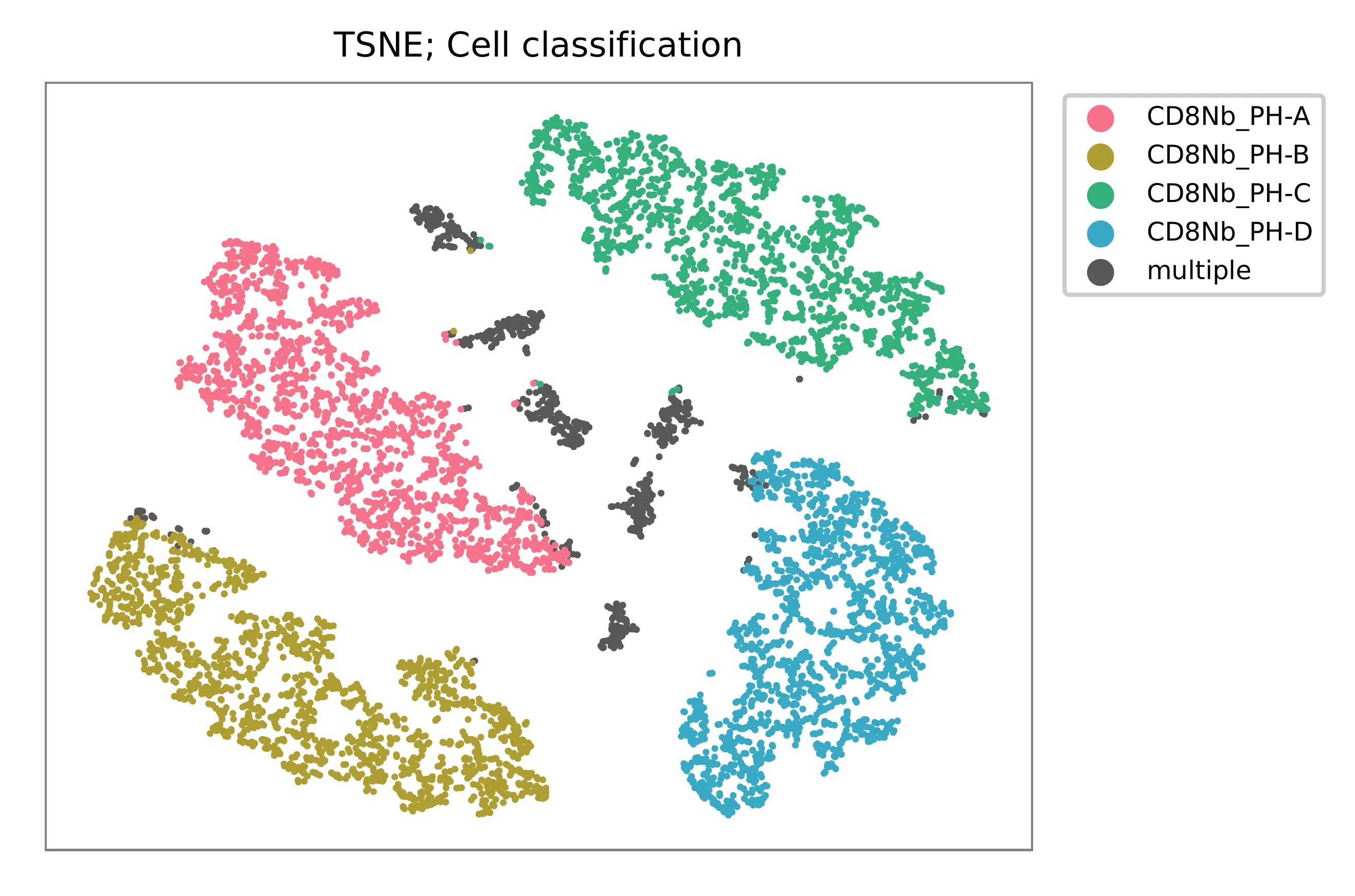
t-SNE embedding of cells based on the abundance of features (phage-derived tags, no transcriptome information used). Colors indicate the hashtag status for each cell, as called by FBA. This is a re-creation of Fig. 3d in Fiskin, E., et al. (2021).
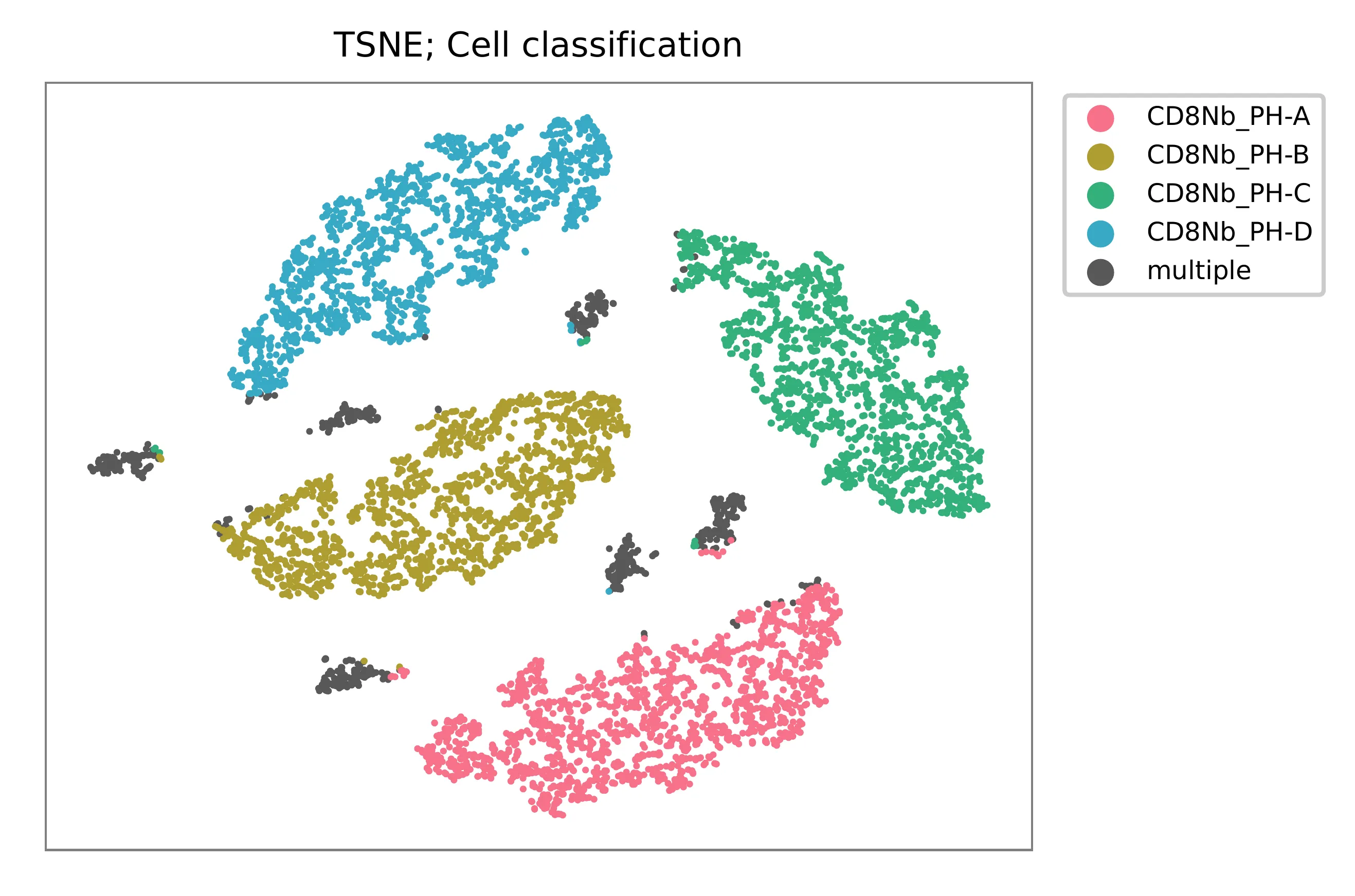
Gaussian mixture model¶
The implementation of demultiplexing method 2 (set by -dm) is
inspired by the method described on the 10x Genomics’ website. To set
the probability threshold for demultiplexing, use -p (default
0.9). To generate visualization plots, set -v.
$ fba demultiplex \
-i matrix_featurecount.csv.gz \
-dm 2 \
-v
2022-03-13 11:27:47,035 - fba.__main__ - INFO - fba version: 0.0.12
2022-03-13 11:27:47,035 - fba.__main__ - INFO - Initiating logging ...
2022-03-13 11:27:47,035 - fba.__main__ - INFO - Python version: 3.9
2022-03-13 11:27:47,035 - fba.__main__ - INFO - Using demultiplex subcommand ...
2022-03-13 11:27:49,515 - fba.__main__ - INFO - Skipping arguments: "-q/--quantile", "-cm/--clustering_method"
2022-03-13 11:27:49,515 - fba.demultiplex - INFO - Output directory: demultiplexed
2022-03-13 11:27:49,515 - fba.demultiplex - INFO - Demultiplexing method: 2
2022-03-13 11:27:49,515 - fba.demultiplex - INFO - UMI normalization method: clr
2022-03-13 11:27:49,515 - fba.demultiplex - INFO - Visualization: On
2022-03-13 11:27:49,515 - fba.demultiplex - INFO - Visualization method: tsne
2022-03-13 11:27:49,515 - fba.demultiplex - INFO - Loading feature count matrix: matrix_featurecount.csv.gz ...
2022-03-13 11:27:49,595 - fba.demultiplex - INFO - Number of cells: 8,366
2022-03-13 11:27:49,595 - fba.demultiplex - INFO - Number of positive cells for a feature to be included: 200
2022-03-13 11:27:49,607 - fba.demultiplex - INFO - Number of features: 4 / 4 (after filtering / original in the matrix)
2022-03-13 11:27:49,607 - fba.demultiplex - INFO - Features: CD8Nb_PH-A CD8Nb_PH-B CD8Nb_PH-C CD8Nb_PH-D
2022-03-13 11:27:49,607 - fba.demultiplex - INFO - Total UMIs: 21,672,447 / 21,672,447
2022-03-13 11:27:49,614 - fba.demultiplex - INFO - Median number of UMIs/reads per cell: 2,261.0 / 2,261.0
2022-03-13 11:27:49,614 - fba.demultiplex - INFO - Demultiplexing ...
2022-03-13 11:27:51,392 - fba.demultiplex - INFO - Generating heatmap ...
2022-03-13 11:27:53,158 - fba.demultiplex - INFO - Embedding ...
2022-03-13 11:28:08,072 - fba.__main__ - INFO - Done.
Heatmap of the relative abundance of features (phage-derived tags, PDTs) across all cells. Each column represents a single cell. This is a re-creation of Fig. 3b in Fiskin, E., et al. (2021).

Preview the demultiplexing result: the numbers of singlets, multiplets and negatives are 6,510 (77.8%), 709 (8.5%), and 1,147 (13.7%), respectively.
In [1]: import pandas as pd
In [2]: m = pd.read_csv("demultiplexed/matrix_cell_identity.csv.gz", index_col=0)
In [3]: m.loc[:, m.sum(axis=0) == 1].sum(axis=1)
Out[3]:
CD8Nb_PH-A 1680
CD8Nb_PH-B 1637
CD8Nb_PH-C 1646
CD8Nb_PH-D 1547
dtype: int64
In [4]: sum(m.sum(axis=0) == 1)
Out[4]: 6510
In [5]: sum(m.sum(axis=0) > 1)
Out[5]: 709
In [6]: sum(m.sum(axis=0) == 0)
Out[6]: 1147
In [7]: m.shape
Out[7]: (4, 8366)
t-SNE embedding of cells based on the abundance of features (phage-derived tags, no transcriptome information used). Colors indicate the hashtag status for each cell, as called by FBA. This is a re-creation of Fig. 3d in Fiskin, E., et al. (2021).
Read distribution and model fitting threshold: