User Guide¶
What is fba?¶
fba is a flexible and streamlined toolbox for quality control,
quantification, demultiplexing of various single-cell feature barcoding
assays. It can be applied to customized feature barcoding
specifications, including different CRISPR constructs or targeted
enriched transcripts. fba allows users to customize a wide range of
parameters for the quantification and demultiplexing process. fba
also has a user-friendly quality control module, which is helpful in
troubleshooting feature barcoding experiments.
Workflow¶
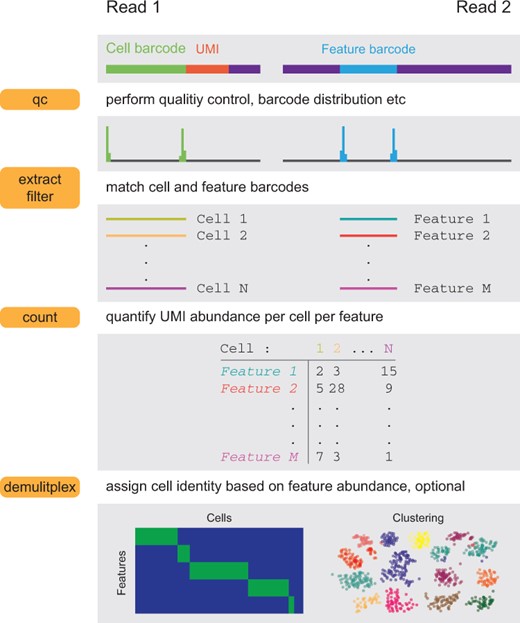
The workflow of FBA: a flexible and streamlined package for single-cell feature barcoding assays.¶
Usage¶
$ fba
usage: fba [-h] ...
Tools for single-cell feature barcoding analysis
optional arguments:
-h, --help show this help message and exit
functions:
extract extract cell and feature barcodes
map map enriched transcripts
filter filter extracted barcodes
count count feature barcodes per cell
demultiplex demultiplex cells based on feature abundance
qc quality control of feature barcoding assay
kallisto_wrapper
deploy kallisto/bustools for feature barcoding
quantification
extract: extract cell and feature barcodes from paired fastq files. For single cell assays, read 1 typically contains cell partitioning and UMI information, while read 2 contains feature information.
map: quantify enriched transcripts (through hybridization or PCR amplification) from parent single cell libraries. Read 1 contains cell partitioning and UMI information, while read 2 contains transcribed regions of enriched/targeted transcripts of interest. BWA (Li, H. 2013) or Bowtie2 (Langmead, B., et al. 2012) is used for read 2 alignment. The quantification (UMI deduplication) of enriched/targeted transcripts is powered by UMI-tools (Smith, T., et al. 2017).
filter: filter extracted cell and feature barcodes (output of
extractorqc). Additional fragment filter/selection can be applied through-cb_seqand/or-fb_seq.count: count UMIs per feature per cell (UMI deduplication), powered by UMI-tools (Smith, T., et al. 2017). The output of
extractorfilteris taken as input.demultiplex: demultiplex cells based on the abundance of features (matrix generated by
countas input).qc: generate diagnostic information. If
-1is omitted, bulk mode is enabled and only read 2 will be analyzed.kallisto_wrapper: deploy kallisto/bustools for feature barcoding quantification (just a wrapper) (Bray, N.L., et al. 2016).